

CRISPR-Cas12a-assisted PCR tagging of mammalian genes
- PMID: 32406907
- PMCID: PMC7265327
- DOI: 10.1083/jcb.201910210
CRISPR-Cas12a-assisted PCR tagging of mammalian genes
Abstract
Here we describe a time-efficient strategy for endogenous C-terminal gene tagging in mammalian tissue culture cells. An online platform is used to design two long gene-specific oligonucleotides for PCR with generic template cassettes to create linear dsDNA donors, termed PCR cassettes. PCR cassettes encode the tag (e.g., GFP), a Cas12a CRISPR RNA for cleavage of the target locus, and short homology arms for directed integration via homologous recombination. The integrated tag is coupled to a generic terminator shielding the tagged gene from the co-inserted auxiliary sequences. Co-transfection of PCR cassettes with a Cas12a-encoding plasmid leads to robust endogenous expression of tagged genes, with tagging efficiency of up to 20% without selection, and up to 60% when selection markers are used. We used target-enrichment sequencing to investigate all potential sources of artifacts. Our work outlines a quick strategy particularly suitable for exploratory studies using endogenous expression of fluorescent protein-tagged genes.
© 2020 Fueller et al.
Figures





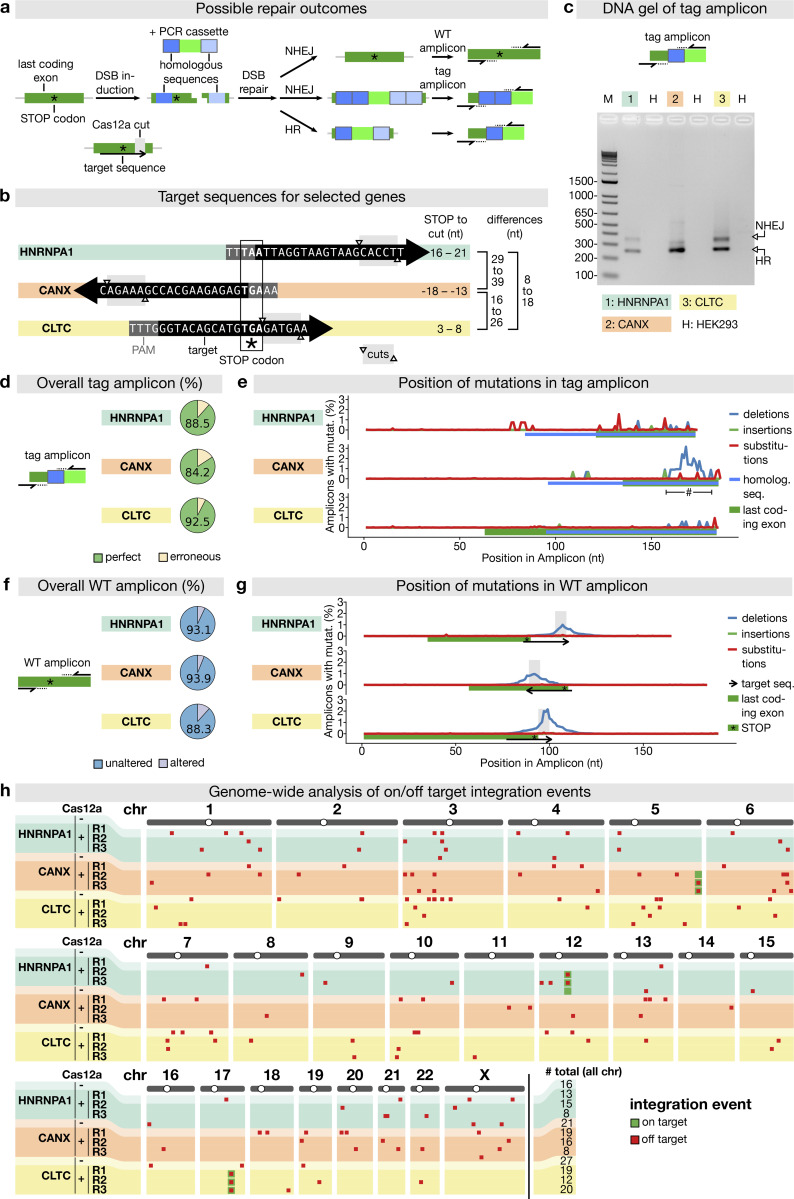







Similar articles
-
Establishment of a Cleavage-Based Single-Plasmid Dual-Luciferase Surrogate Reporter for the Cleavage Efficiency Evaluation of CRISPR-Cas12a Systems and Its Primary Application.CRISPR J. 2024 Jun;7(3):156-167. doi: 10.1089/crispr.2024.0038. CRISPR J. 2024. PMID: 38922054
-
An Isothermal Method for Sensitive Detection of Mycobacterium tuberculosis Complex Using Clustered Regularly Interspaced Short Palindromic Repeats/Cas12a Cis and Trans Cleavage.J Mol Diagn. 2020 Aug;22(8):1020-1029. doi: 10.1016/j.jmoldx.2020.04.212. Epub 2020 May 26. J Mol Diagn. 2020. PMID: 32470556
-
A Survey of Genome Editing Activity for 16 Cas12a Orthologs.Keio J Med. 2020 Sep 25;69(3):59-65. doi: 10.2302/kjm.2019-0009-OA. Epub 2019 Nov 14. Keio J Med. 2020. PMID: 31723075 Free PMC article.
-
CRISPR-Cas12a System for Biosensing and Gene Regulation.Chem Asian J. 2021 Apr 19;16(8):857-867. doi: 10.1002/asia.202100043. Epub 2021 Mar 18. Chem Asian J. 2021. PMID: 33638271 Review.
-
Editor's cut: DNA cleavage by CRISPR RNA-guided nucleases Cas9 and Cas12a.Biochem Soc Trans. 2020 Feb 28;48(1):207-219. doi: 10.1042/BST20190563. Biochem Soc Trans. 2020. PMID: 31872209 Free PMC article. Review.
Cited by
-
qTAG: an adaptable plasmid scaffold for CRISPR-based endogenous tagging.EMBO J. 2025 Feb;44(3):947-974. doi: 10.1038/s44318-024-00337-5. Epub 2024 Dec 12. EMBO J. 2025. PMID: 39668248 Free PMC article.
-
Constitutional PIGA mutations cause a novel subtype of hemochromatosis in patients with neurologic dysfunction.Blood. 2022 Mar 3;139(9):1418-1422. doi: 10.1182/blood.2021013519. Blood. 2022. PMID: 34875027 Free PMC article. No abstract available.
-
Pooled clone collections by multiplexed CRISPR-Cas12a-assisted gene tagging in yeast.Nat Commun. 2019 Jul 4;10(1):2960. doi: 10.1038/s41467-019-10816-7. Nat Commun. 2019. PMID: 31273196 Free PMC article.
-
Triple-Branch Catalytic Assembly DNAzyme Motivated DNA Tweezer for Sensitive and Reliable mecA Gene Detection in Staphylococcus aureus.J Microbiol Biotechnol. 2024 Dec 28;34(12):2450-2456. doi: 10.4014/jmb.2409.09008. Epub 2024 Oct 1. J Microbiol Biotechnol. 2024. PMID: 39467691 Free PMC article.
-
Rhomboid protease RHBDL4 promotes retrotranslocation of aggregation-prone proteins for degradation.Cell Rep. 2022 Aug 9;40(6):111175. doi: 10.1016/j.celrep.2022.111175. Cell Rep. 2022. PMID: 35947953 Free PMC article.
References
-
- Attali D. 2017. shinyjs: Easily Improve the User Experience of Your Shiny Apps in Seconds. R package version 1.0. https://cran.r-project.org/web/packages/shinyjs/index.html
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
