

Dynamic landscape and evolution of m6A methylation in human
- PMID: 32406913
- PMCID: PMC7293016
- DOI: 10.1093/nar/gkaa347
Dynamic landscape and evolution of m6A methylation in human
Abstract
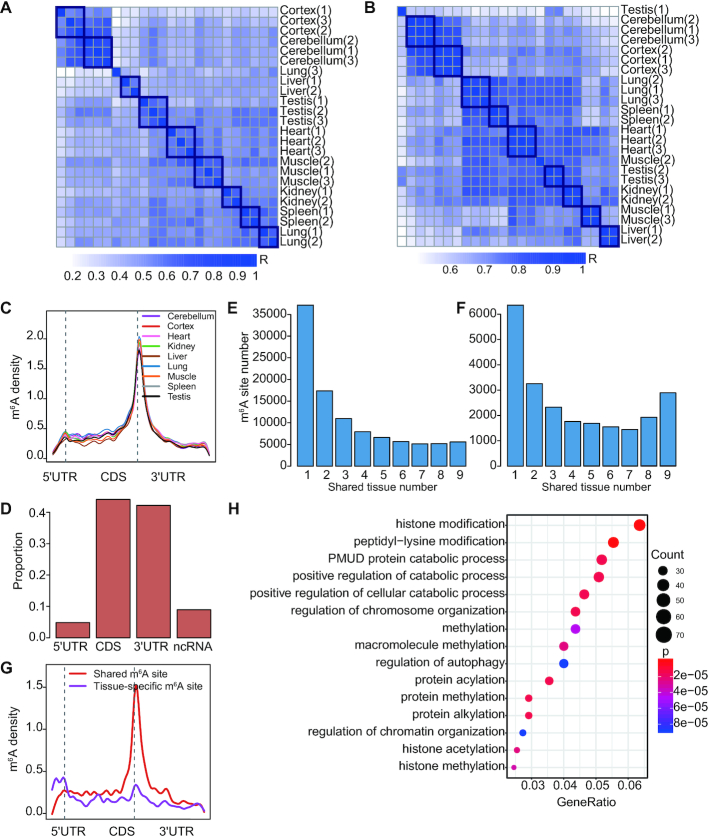
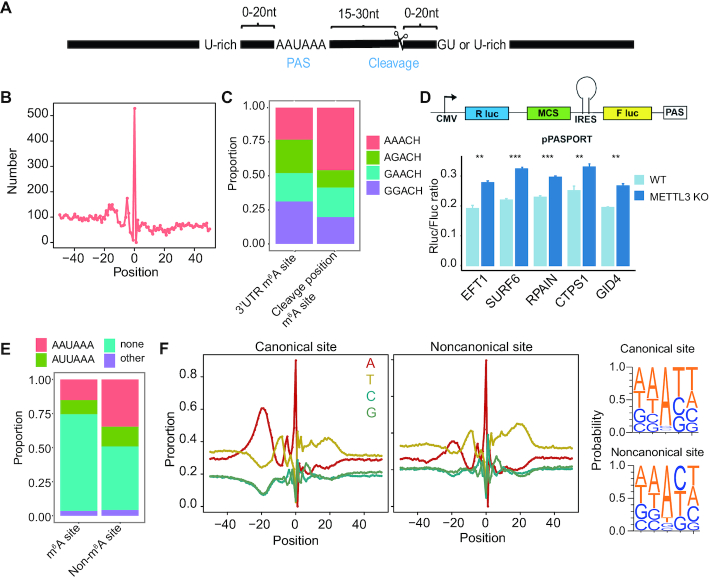
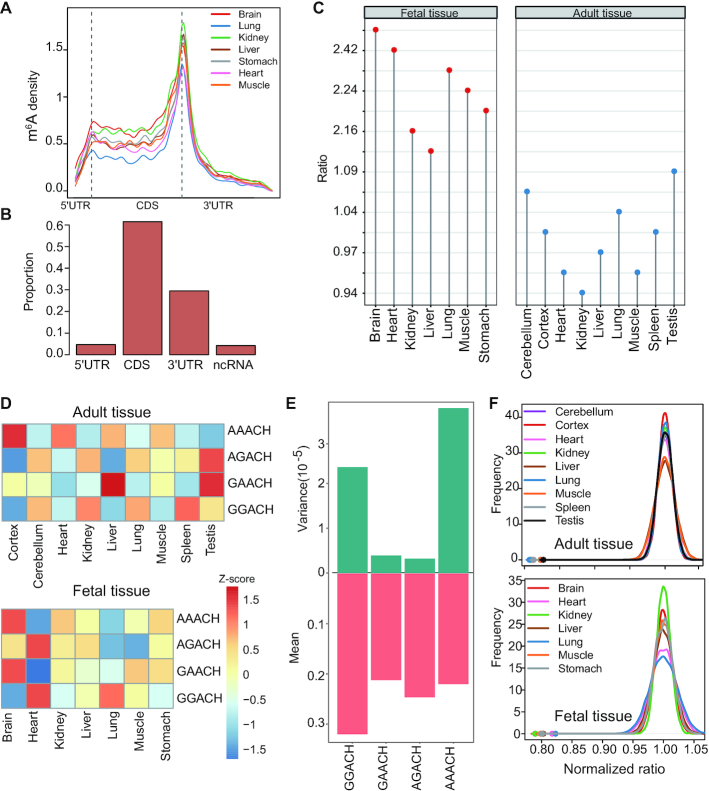
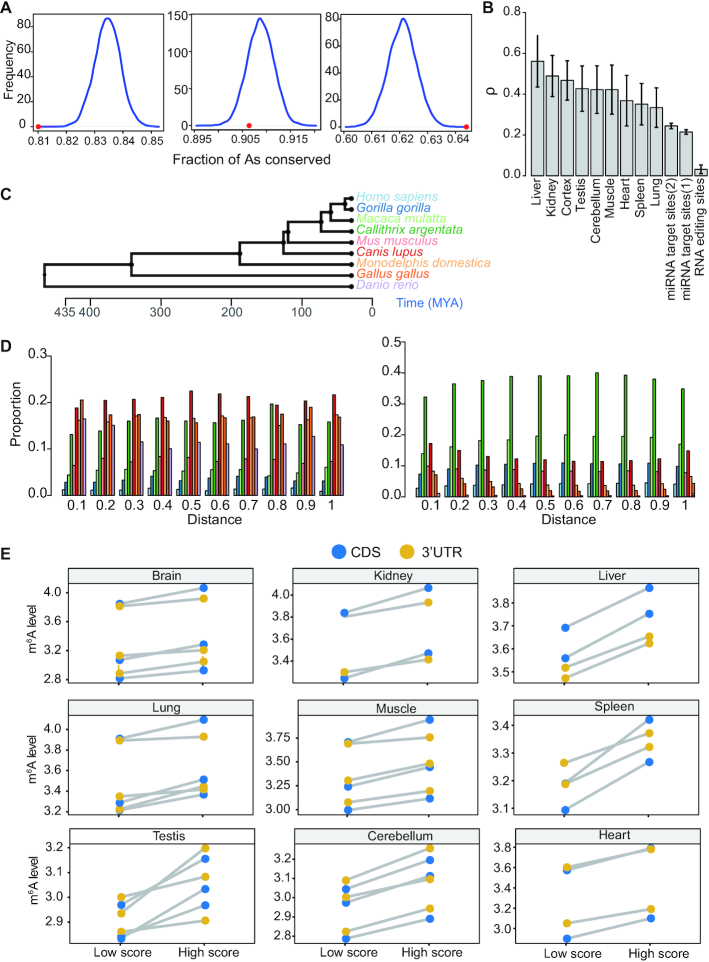
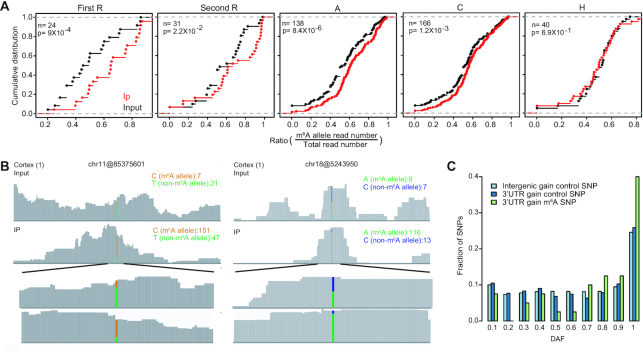
m6A is a prevalent internal modification in mRNAs and has been linked to the diverse effects on mRNA fate. To explore the landscape and evolution of human m6A, we generated 27 m6A methylomes across major adult tissues. These data reveal dynamic m6A methylation across tissue types, uncover both broadly or tissue-specifically methylated sites, and identify an unexpected enrichment of m6A methylation at non-canonical cleavage sites. A comparison of fetal and adult m6A methylomes reveals that m6A preferentially occupies CDS regions in fetal tissues. Moreover, the m6A sub-motifs vary between fetal and adult tissues or across tissue types. From the evolutionary perspective, we uncover that the selection pressure on m6A sites varies and depends on their genic locations. Unexpectedly, we found that ∼40% of the 3'UTR m6A sites are under negative selection, which is higher than the evolutionary constraint on miRNA binding sites, and much higher than that on A-to-I RNA modification. Moreover, the recently gained m6A sites in human populations are clearly under positive selection and associated with traits or diseases. Our work provides a resource of human m6A profile for future studies of m6A functions, and suggests a role of m6A modification in human evolutionary adaptation and disease susceptibility.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Li S., Mason C.E.. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 2014; 15:127–150. - PubMed
-
- Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. et al.. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201–206. - PubMed
-
- Li X., Xiong X., Yi C.. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods. 2016; 14:23–31. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
