

Mutation-Directed Therapeutics for Neurofibromatosis Type I
- PMID: 32408052
- PMCID: PMC7225739
- DOI: 10.1016/j.omtn.2020.04.012
Mutation-Directed Therapeutics for Neurofibromatosis Type I
Abstract
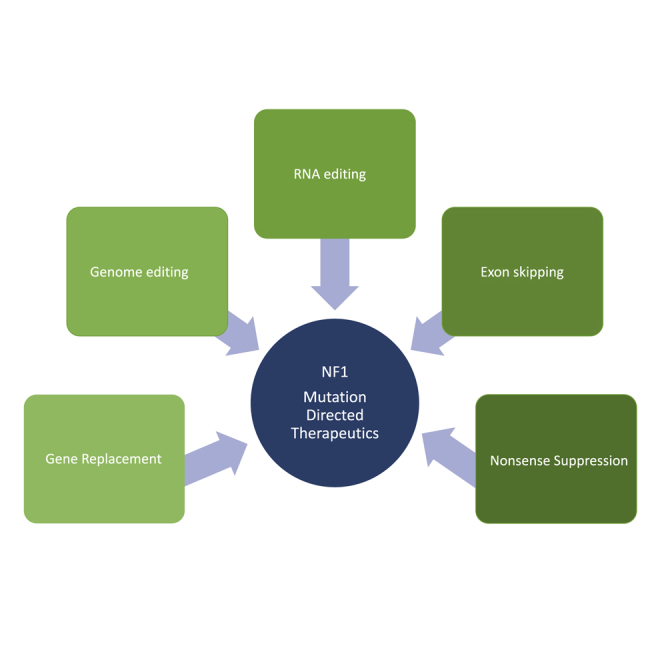
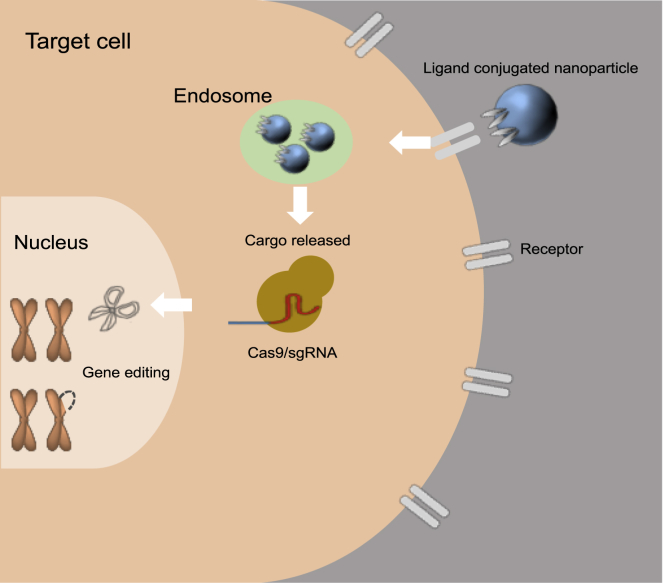
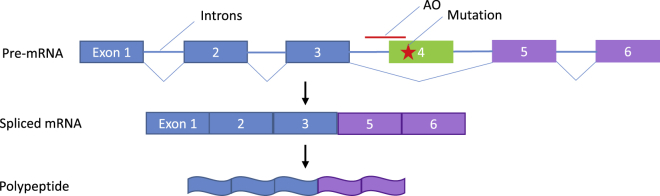
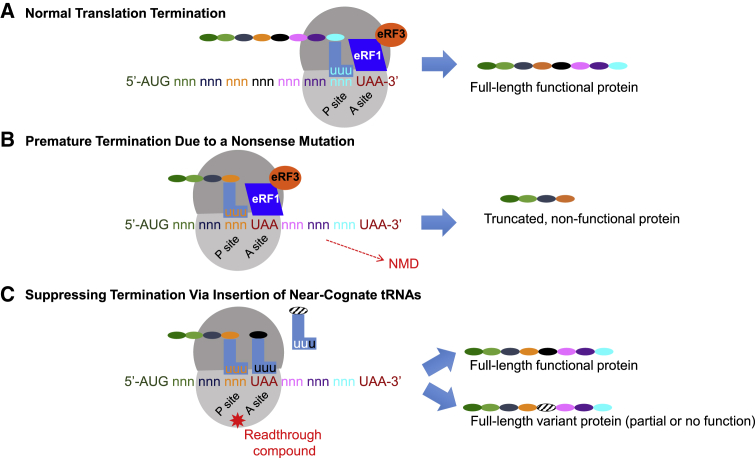
Significant advances in biotechnology have led to the development of a number of different mutation-directed therapies. Some of these techniques have matured to a level that has allowed testing in clinical trials, but few have made it to approval by drug-regulatory bodies for the treatment of specific diseases. While there are still various hurdles to be overcome, recent success stories have proven the potential power of mutation-directed therapies and have fueled the hope of finding therapeutics for other genetic disorders. In this review, we summarize the state-of-the-art of various therapeutic approaches and assess their applicability to the genetic disorder neurofibromatosis type I (NF1). NF1 is caused by the loss of function of neurofibromin, a tumor suppressor and downregulator of the Ras signaling pathway. The condition is characterized by a variety of phenotypes and includes symptoms such as skin spots, nervous system tumors, skeletal dysplasia, and others. Hence, depending on the patient, therapeutics may need to target different tissues and cell types. While we also discuss the delivery of therapeutics, in particular via viral vectors and nanoparticles, our main focus is on therapeutic techniques that reconstitute functional neurofibromin, most notably cDNA replacement, CRISPR-based DNA repair, RNA repair, antisense oligonucleotide therapeutics including exon skipping, and nonsense suppression.
Copyright © 2020. Published by Elsevier Inc.
Figures

References
-
- Friedman J.M., Gutmann D.H., MacCollin M., Riccardi V.M., editors. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Third Edition. Johns Hopkins University Press; 1999.
-
- Kallionpää R.A., Uusitalo E., Leppävirta J., Pöyhönen M., Peltonen S., Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018;20:1082–1086. - PubMed
-
- Buratti E., Baralle D. Exon skipping mutations in neurofibromatosis. Methods Mol. Biol. 2012;867:65–76. - PubMed
-
- Thomson S.A., Wallace M.R. RT-PCR splicing analysis of the NF1 open reading frame. Hum. Genet. 2002;110:495–502. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
