

Proteomics of SARS-CoV-2-infected host cells reveals therapy targets
- PMID: 32408336
- PMCID: PMC7616921
- DOI: 10.1038/s41586-020-2332-7
Proteomics of SARS-CoV-2-infected host cells reveals therapy targets
Abstract
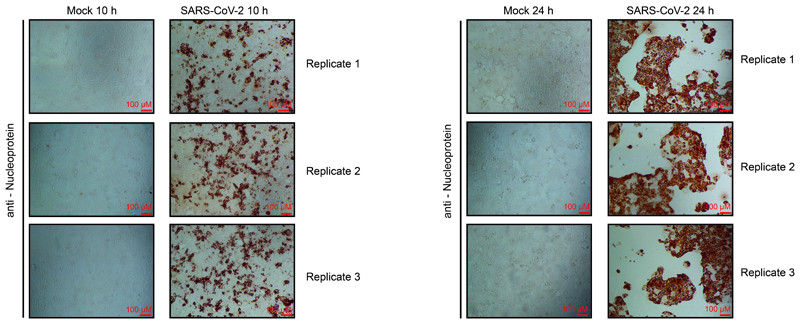
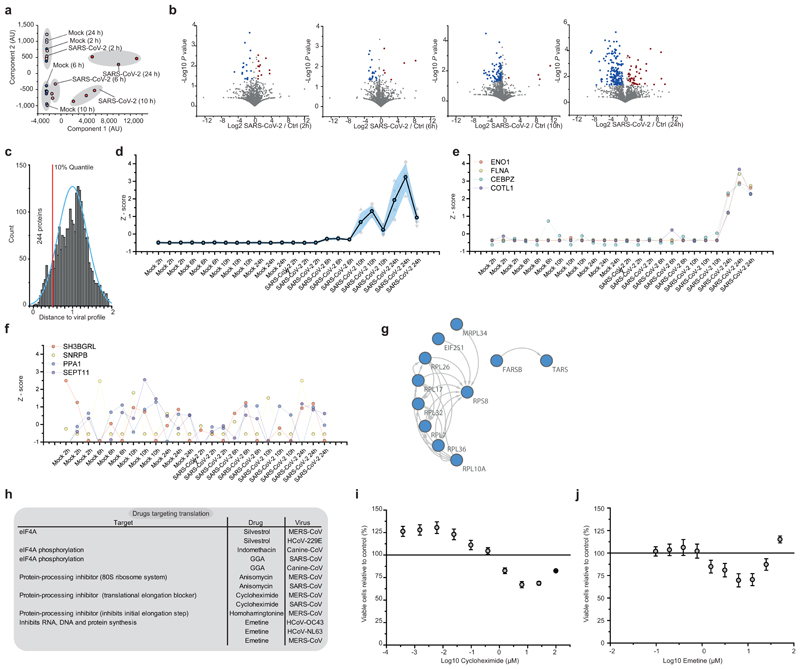
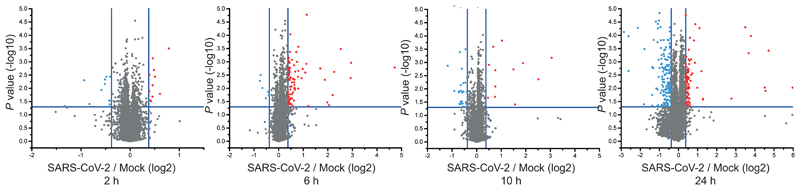
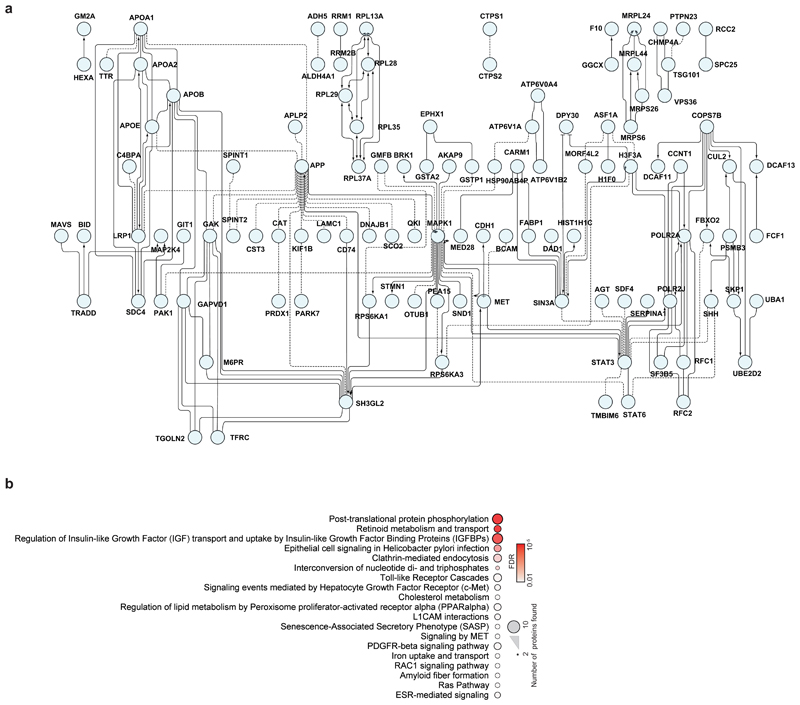
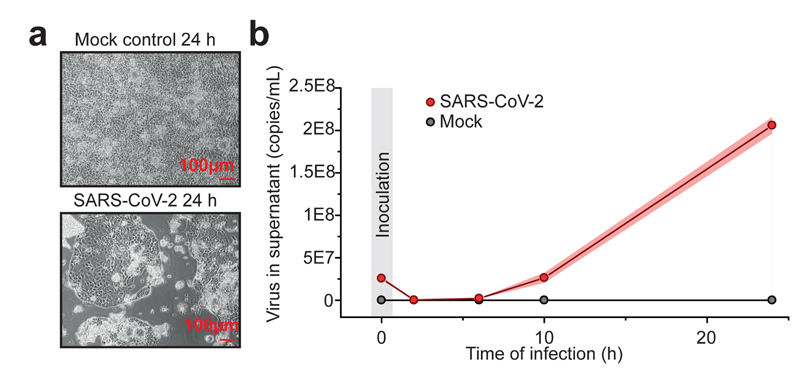
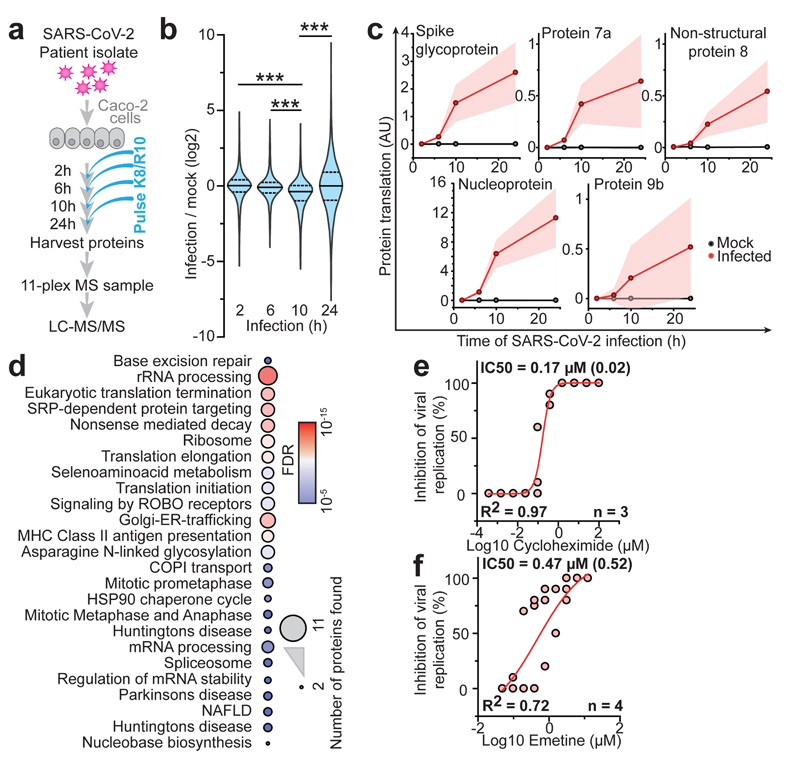
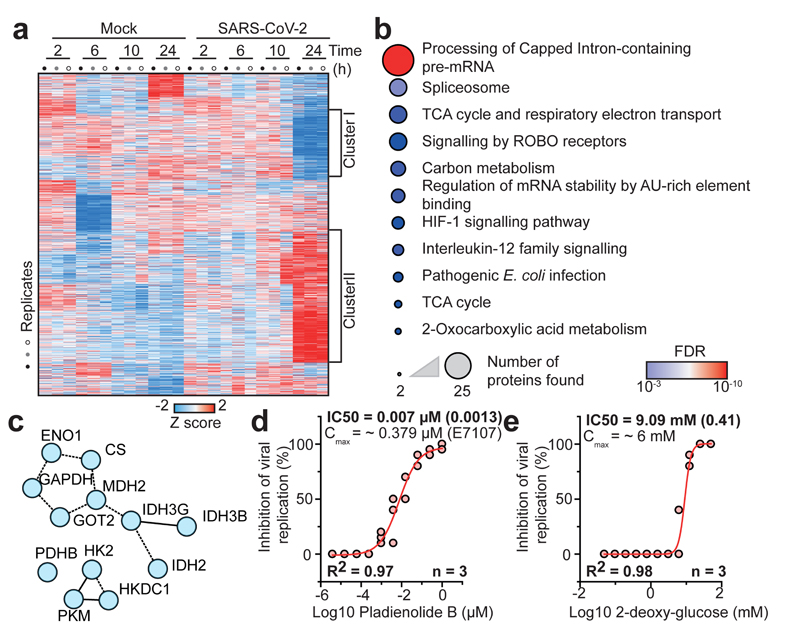
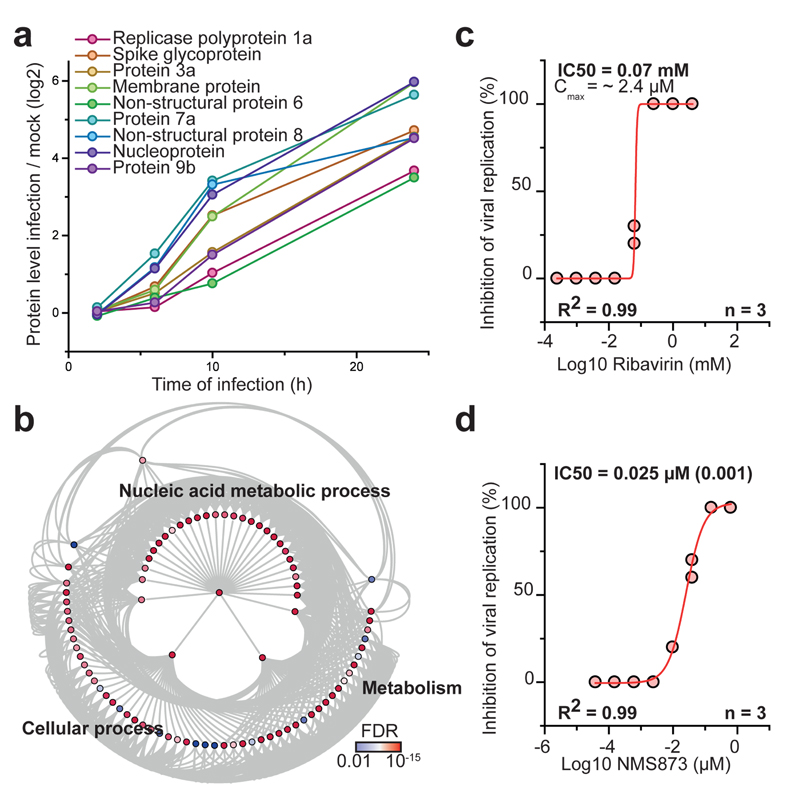
A new coronavirus was recently discovered and named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Infection with SARS-CoV-2 in humans causes coronavirus disease 2019 (COVID-19) and has been rapidly spreading around the globe1,2. SARS-CoV-2 shows some similarities to other coronaviruses; however, treatment options and an understanding of how SARS-CoV-2 infects cells are lacking. Here we identify the host cell pathways that are modulated by SARS-CoV-2 and show that inhibition of these pathways prevents viral replication in human cells. We established a human cell-culture model for infection with a clinical isolate of SARS-CoV-2. Using this cell-culture system, we determined the infection profile of SARS-CoV-2 by translatome3 and proteome proteomics at different times after infection. These analyses revealed that SARS-CoV-2 reshapes central cellular pathways such as translation, splicing, carbon metabolism, protein homeostasis (proteostasis) and nucleic acid metabolism. Small-molecule inhibitors that target these pathways prevented viral replication in cells. Our results reveal the cellular infection profile of SARS-CoV-2 and have enabled the identification of drugs that inhibit viral replication. We anticipate that our results will guide efforts to understand the molecular mechanisms that underlie the modulation of host cells after infection with SARS-CoV-2. Furthermore, our findings provide insights for the development of therapies for the treatment of COVID-19.
Conflict of interest statement
The authors declare no competing interests.
Figures
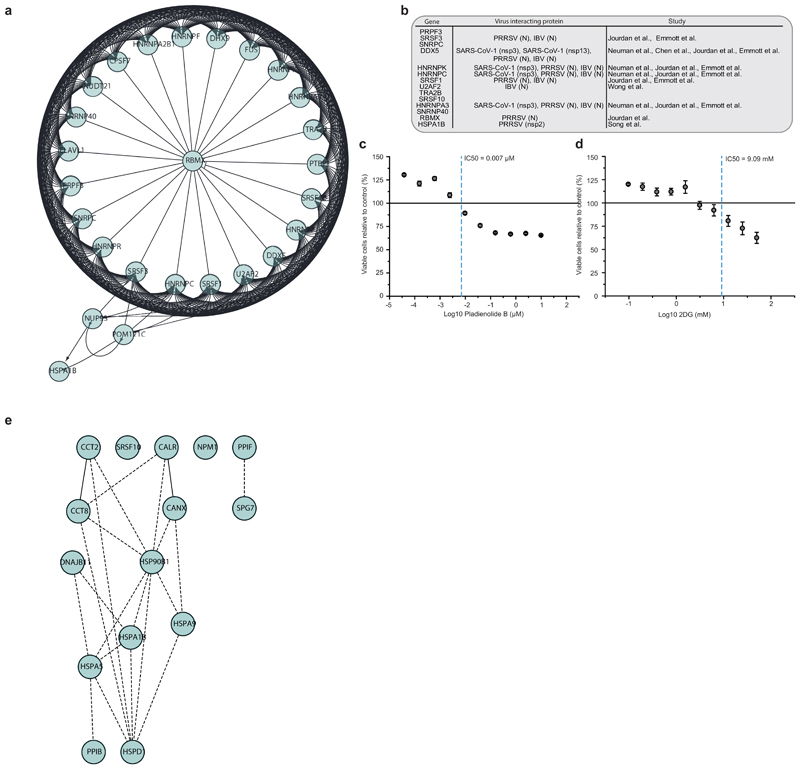
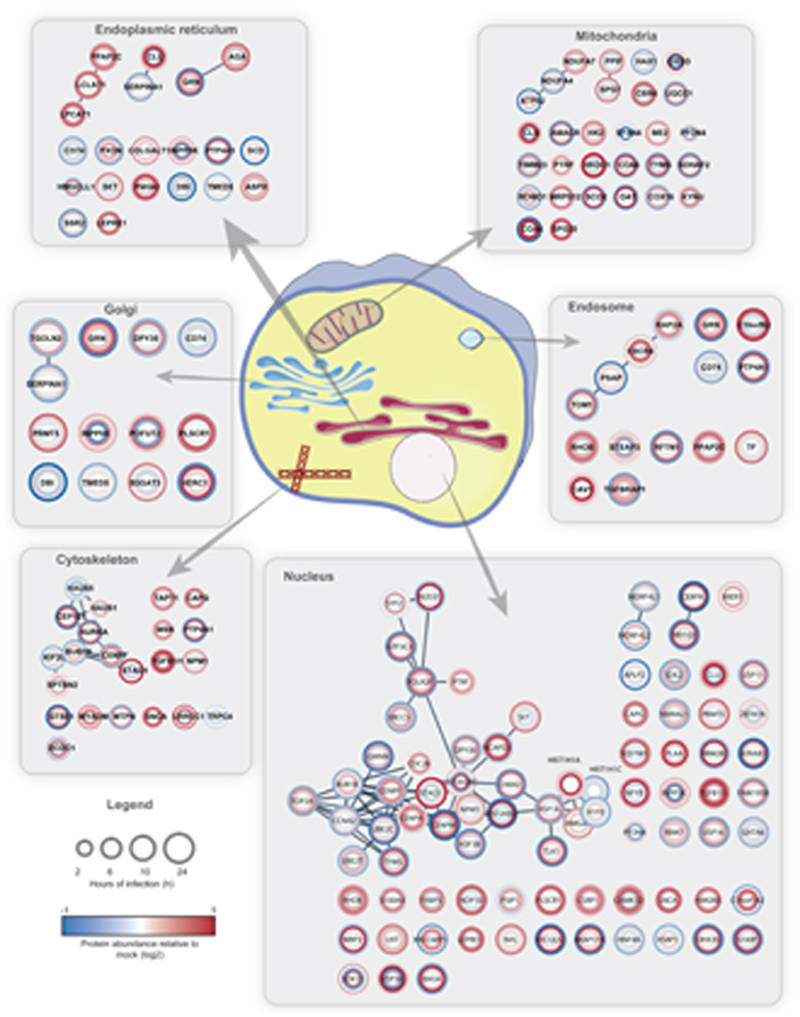
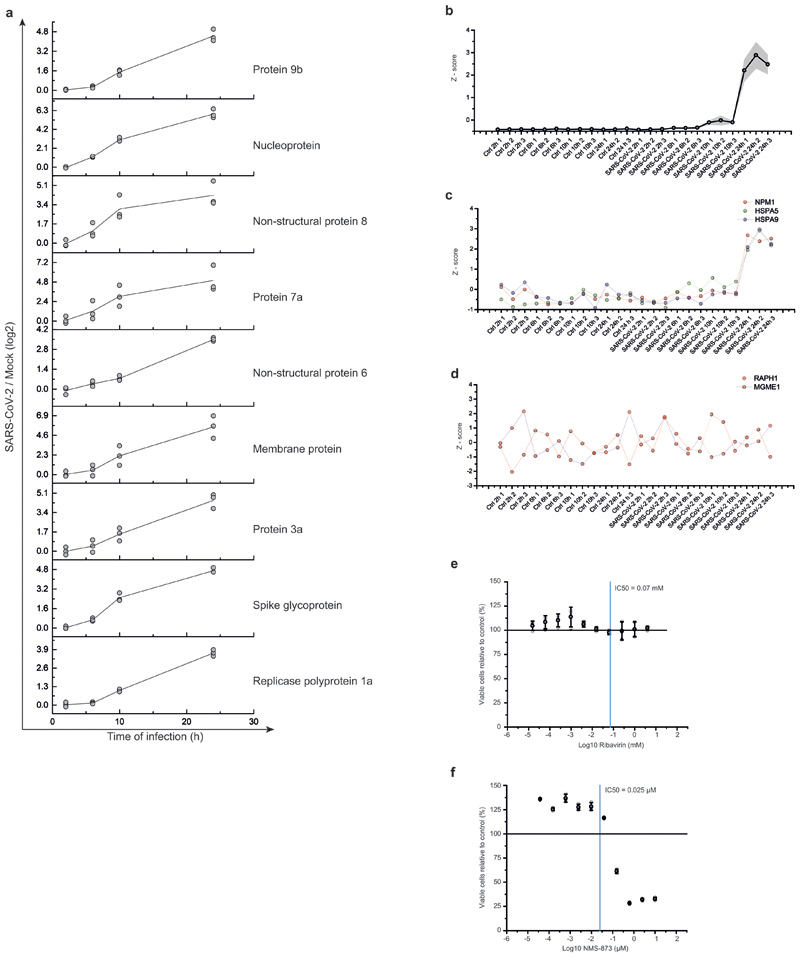
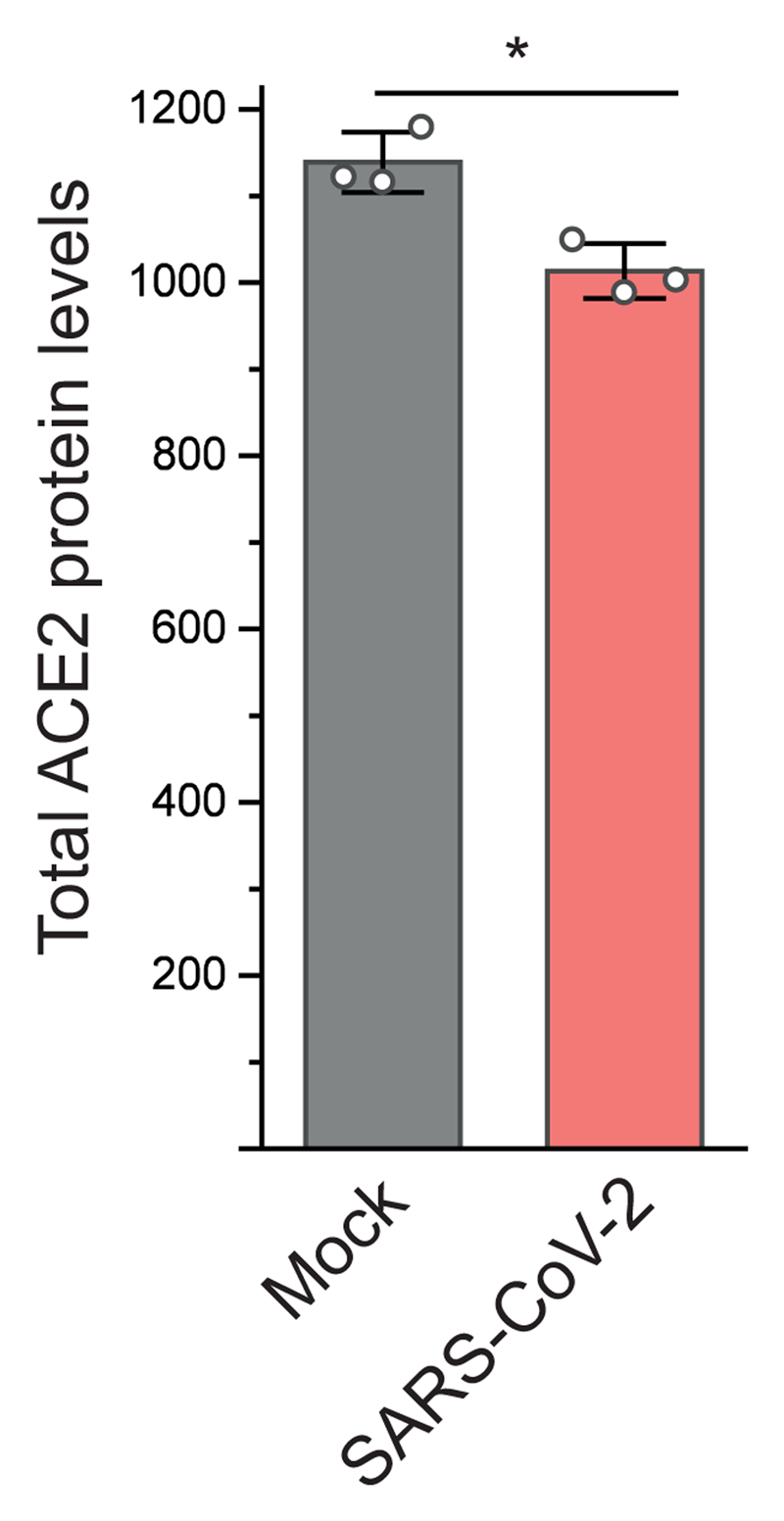
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
