

Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures
- PMID: 32409712
- PMCID: PMC7224215
- DOI: 10.1038/s41467-020-16212-w
Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures
Abstract
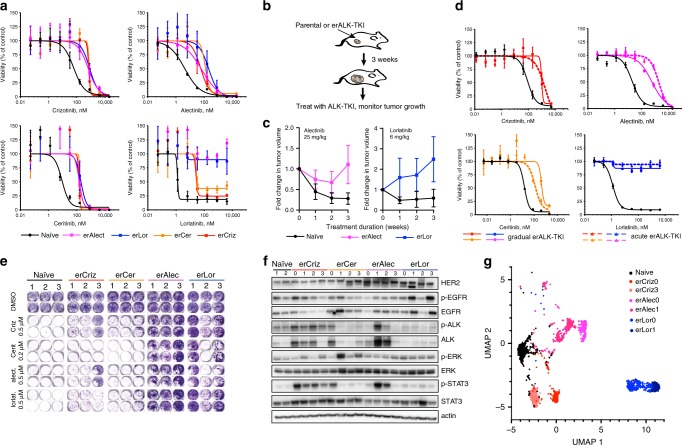
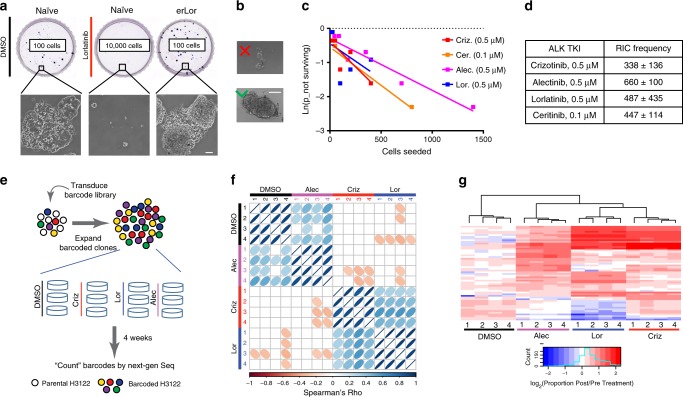
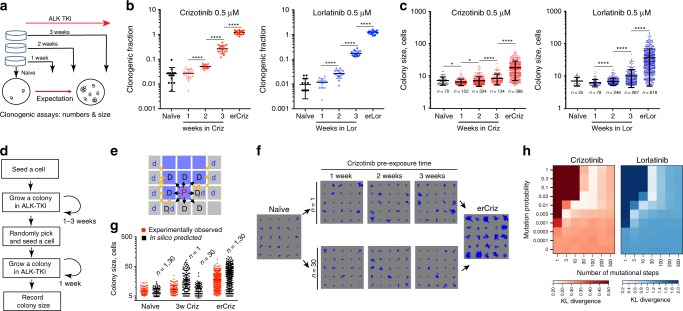
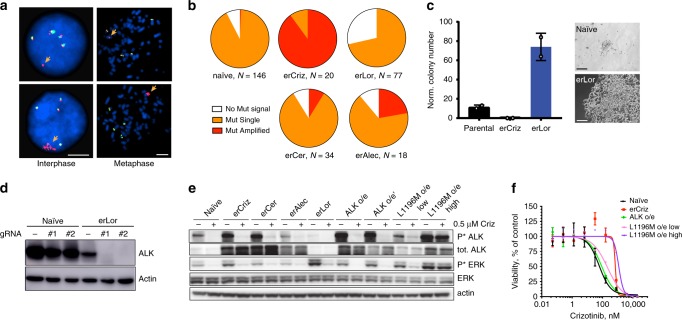
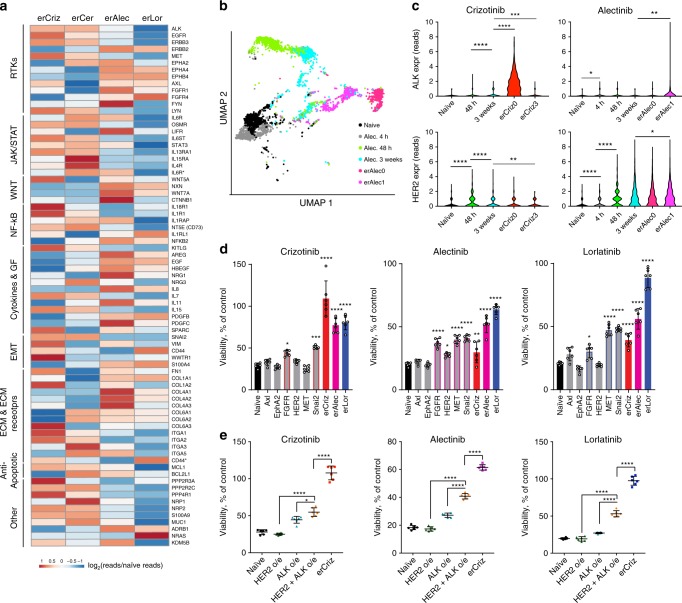
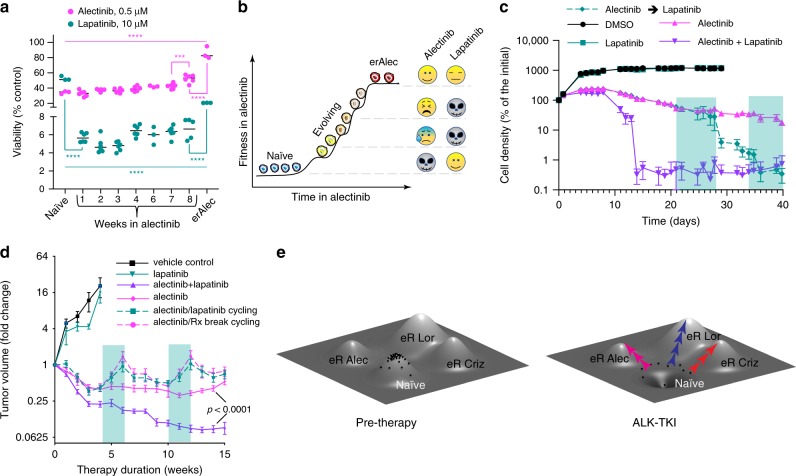
Despite high initial efficacy, targeted therapies eventually fail in advanced cancers, as tumors develop resistance and relapse. In contrast to the substantial body of research on the molecular mechanisms of resistance, understanding of how resistance evolves remains limited. Using an experimental model of ALK positive NSCLC, we explored the evolution of resistance to different clinical ALK inhibitors. We found that resistance can originate from heterogeneous, weakly resistant subpopulations with variable sensitivity to different ALK inhibitors. Instead of the commonly assumed stochastic single hit (epi) mutational transition, or drug-induced reprogramming, we found evidence for a hybrid scenario involving the gradual, multifactorial adaptation to the inhibitors through acquisition of multiple cooperating genetic and epigenetic adaptive changes. Additionally, we found that during this adaptation tumor cells might present unique, temporally restricted collateral sensitivities, absent in therapy naïve or fully resistant cells, suggesting the potential for new therapeutic interventions, directed against evolving resistance.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
