

An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells
- PMID: 32413320
- PMCID: PMC7529102
- DOI: 10.1016/j.cell.2020.04.048
An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells
Erratum in
-
An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells.Cell. 2020 Jun 25;181(7):1693-1694. doi: 10.1016/j.cell.2020.06.018. Cell. 2020. PMID: 32589959 Free PMC article. No abstract available.
Abstract
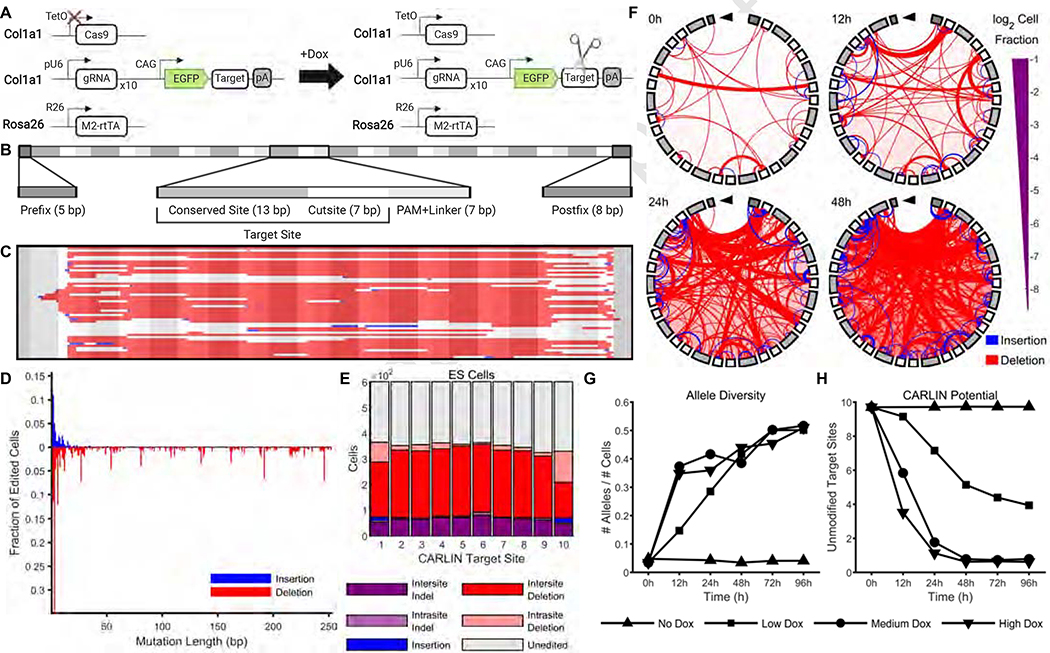
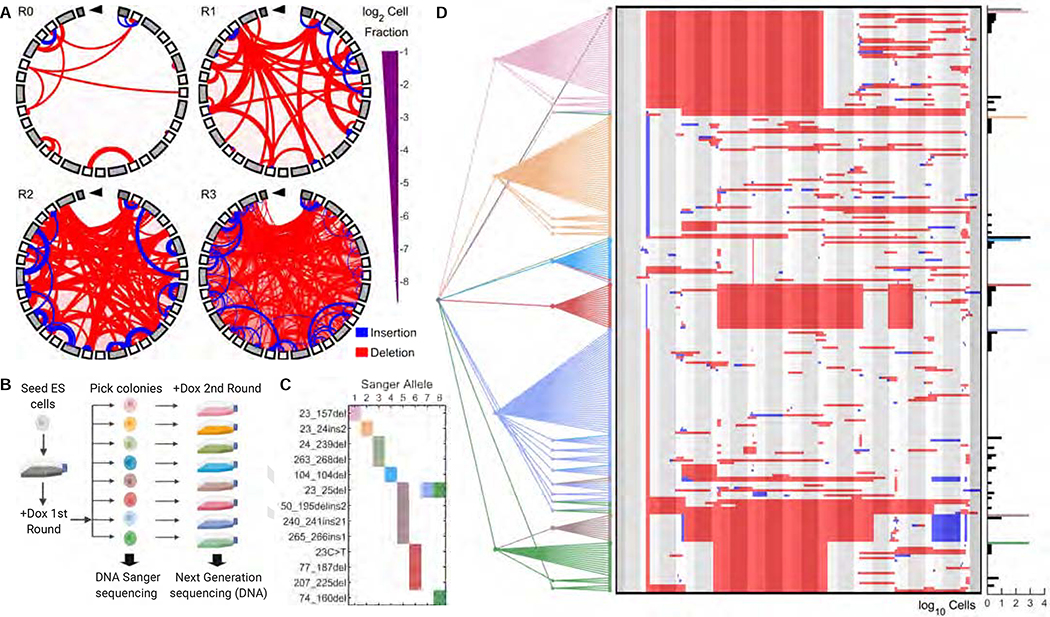
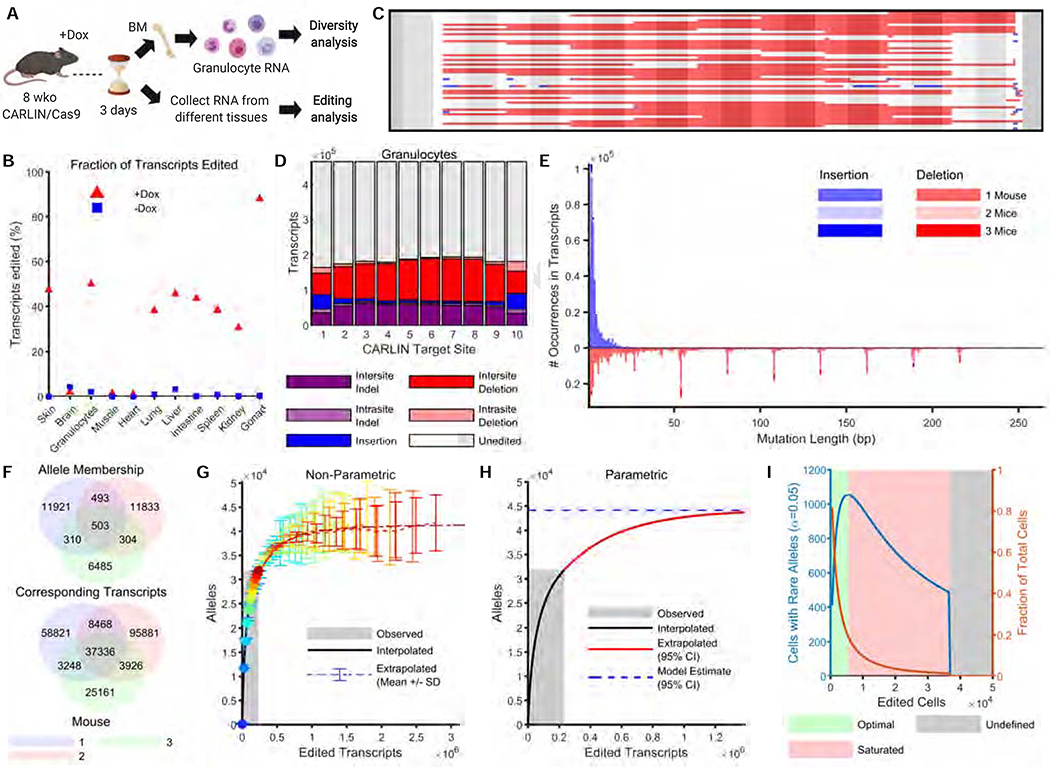
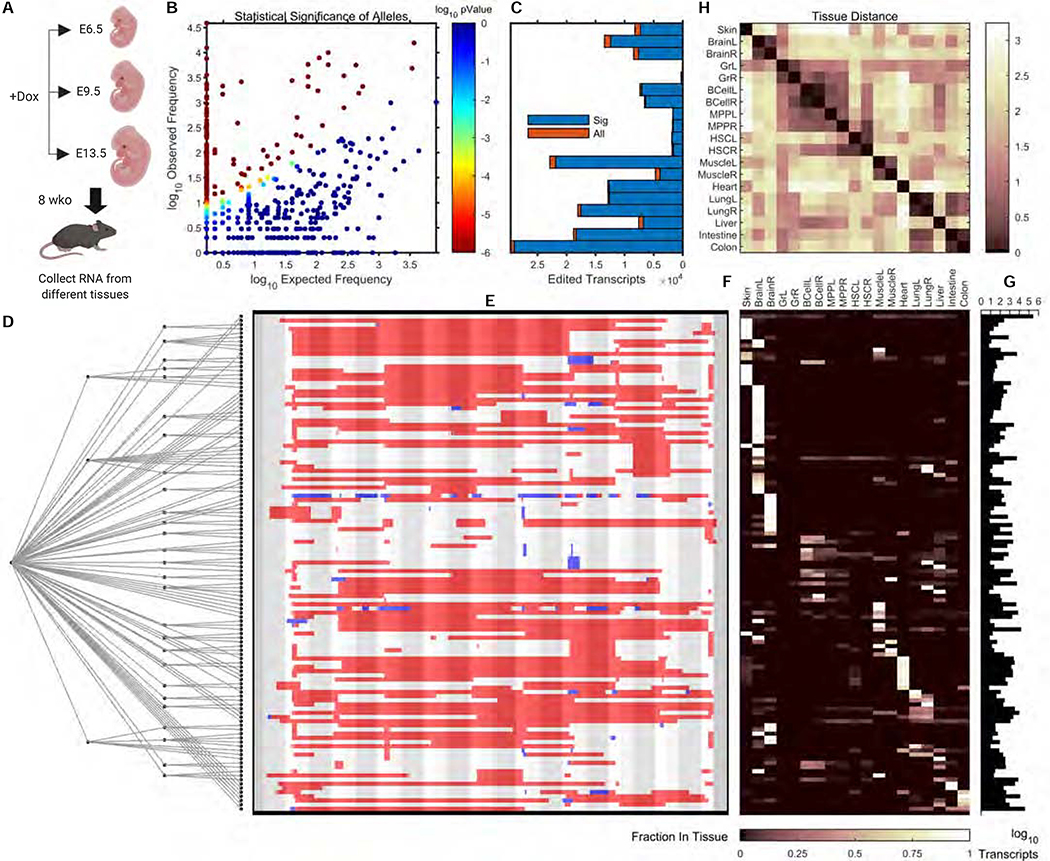
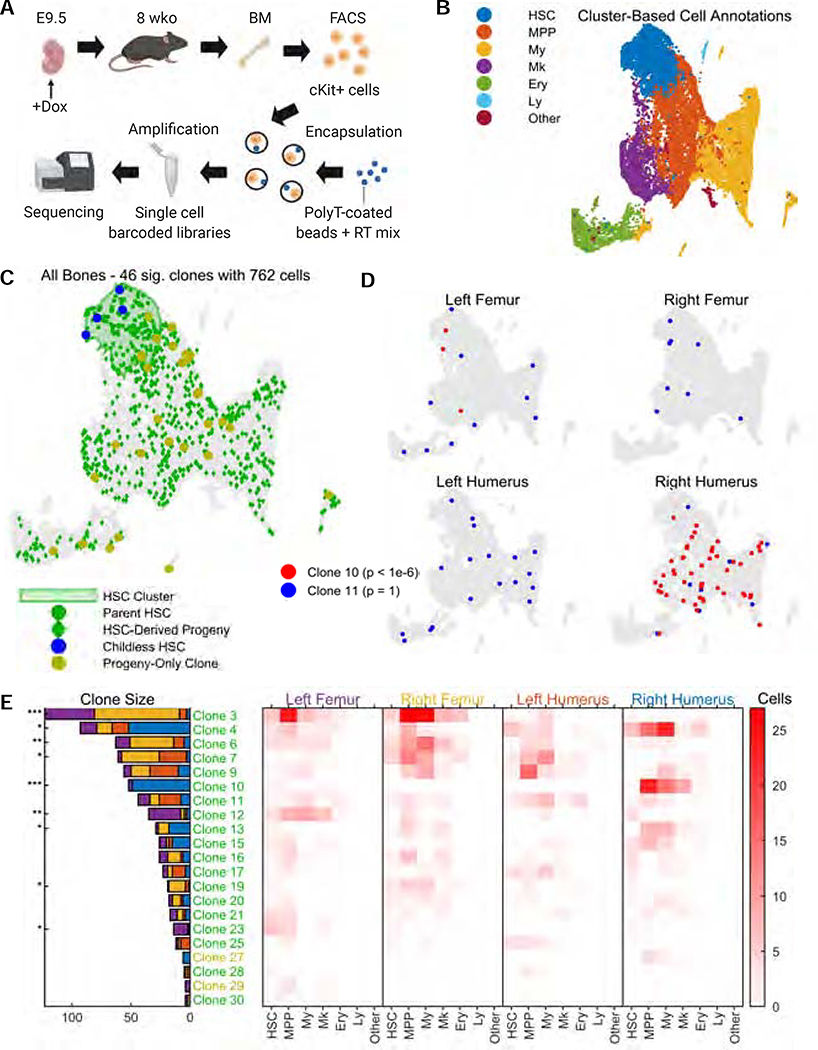
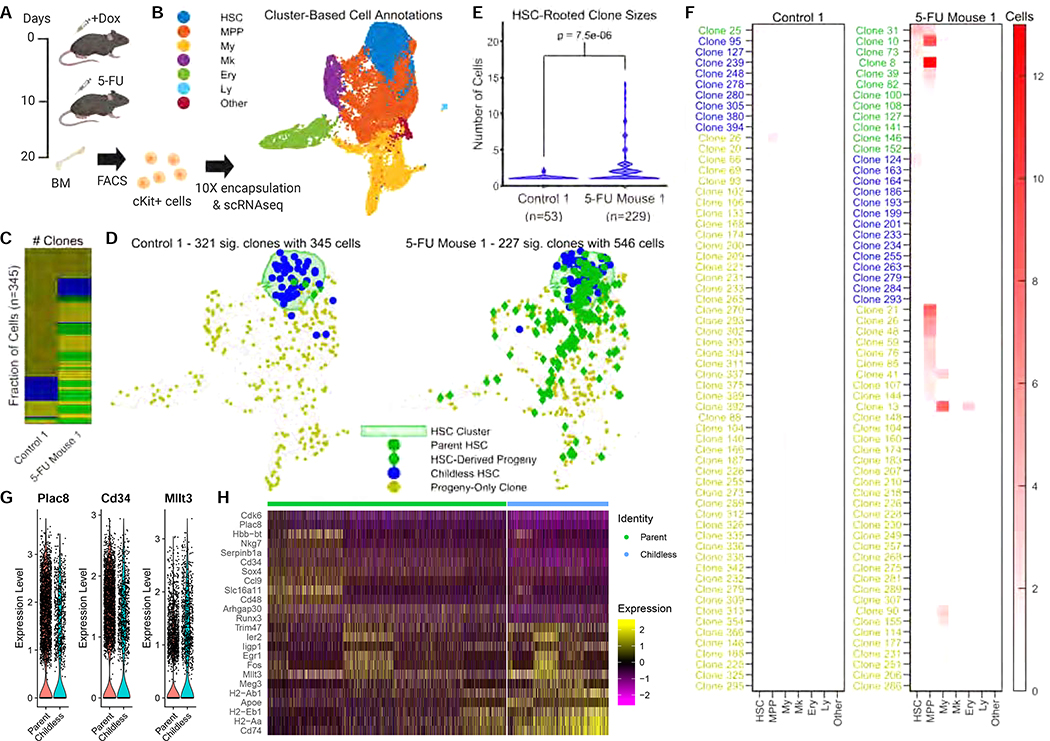
Tracing the lineage history of cells is key to answering diverse and fundamental questions in biology. Coupling of cell ancestry information with other molecular readouts represents an important goal in the field. Here, we describe the CRISPR array repair lineage tracing (CARLIN) mouse line and corresponding analysis tools that can be used to simultaneously interrogate the lineage and transcriptomic information of single cells in vivo. This model exploits CRISPR technology to generate up to 44,000 transcribed barcodes in an inducible fashion at any point during development or adulthood, is compatible with sequential barcoding, and is fully genetically defined. We have used CARLIN to identify intrinsic biases in the activity of fetal liver hematopoietic stem cell (HSC) clones and to uncover a previously unappreciated clonal bottleneck in the response of HSCs to injury. CARLIN also allows the unbiased identification of transcriptional signatures associated with HSC activity without cell sorting.
Keywords: barcoding; hematopoiesis; lineage tracing; single cell; stem cells.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Comment in
-
A mouse with history.Nat Rev Genet. 2020 Jul;21(7):388. doi: 10.1038/s41576-020-0252-x. Nat Rev Genet. 2020. PMID: 32457510 No abstract available.
References
-
- Alemany A, Florescu M, Baron CS, Peterson-Maduro J and Van Oudenaarden A (2018). Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112. - PubMed
-
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, Wilcox S, Tai L, Strasser A, and Herold MJ (2015). An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep 10, 1422–1432 - PubMed
-
- Balakier H and Pedersen RA (1982). Allocation of cells to inner cell mass and trophectoderm lineages in preimplantation mouse embryos. Dev. Biol. 90, 352–362. - PubMed
-
- Beard C, Hochedlinger K, Plath K, Wutz A and Jaenisch R (2006). Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28. - PubMed
-
- Buchholz F, Angrand PO, and Stewart AF (1998). Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol 16, 657–662. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
