

Psi4 1.4: Open-source software for high-throughput quantum chemistry
- PMID: 32414239
- PMCID: PMC7228781
- DOI: 10.1063/5.0006002
Psi4 1.4: Open-source software for high-throughput quantum chemistry
Abstract
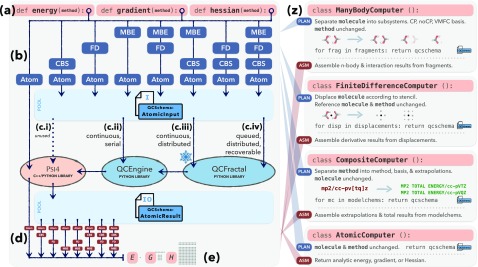
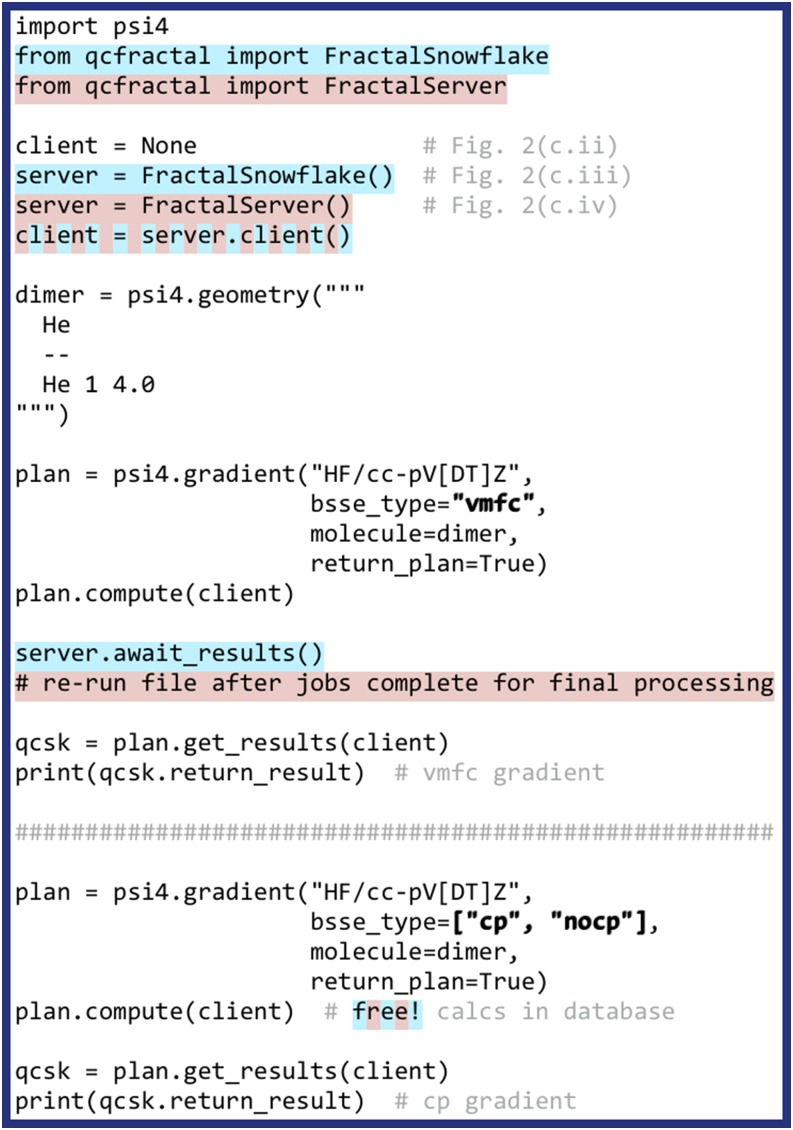
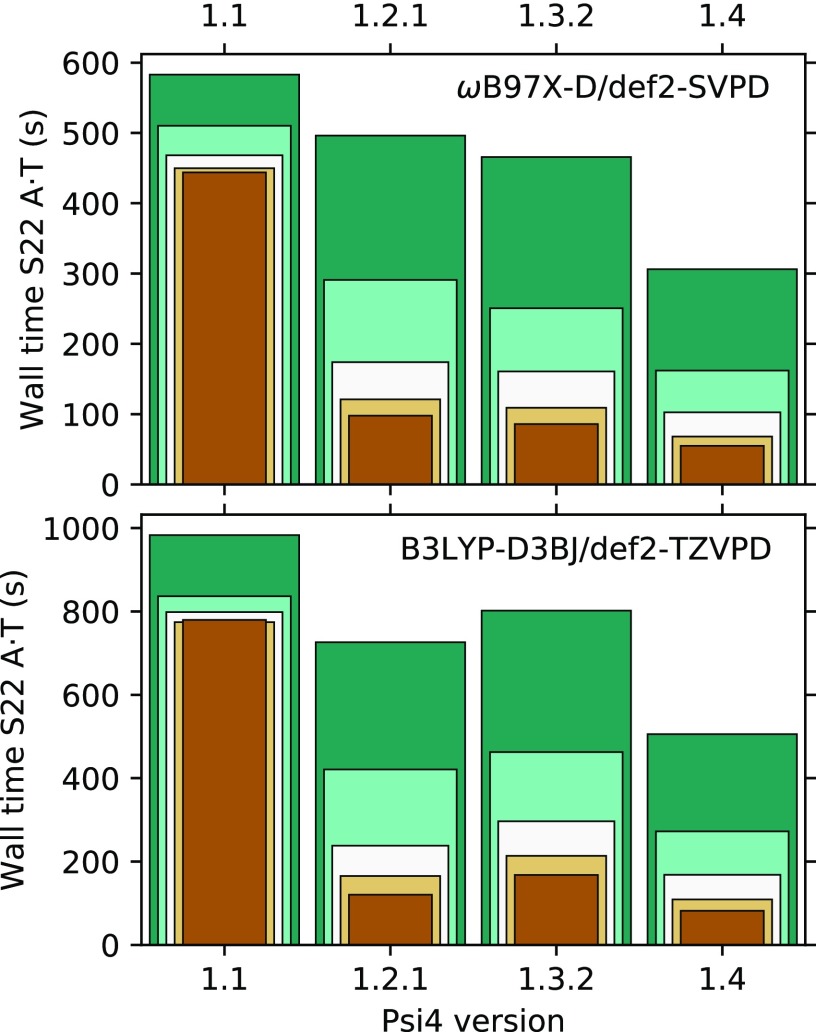
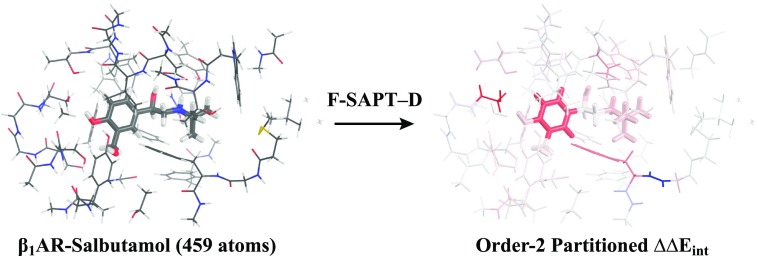
PSI4 is a free and open-source ab initio electronic structure program providing implementations of Hartree-Fock, density functional theory, many-body perturbation theory, configuration interaction, density cumulant theory, symmetry-adapted perturbation theory, and coupled-cluster theory. Most of the methods are quite efficient, thanks to density fitting and multi-core parallelism. The program is a hybrid of C++ and Python, and calculations may be run with very simple text files or using the Python API, facilitating post-processing and complex workflows; method developers also have access to most of PSI4's core functionalities via Python. Job specification may be passed using The Molecular Sciences Software Institute (MolSSI) QCSCHEMA data format, facilitating interoperability. A rewrite of our top-level computation driver, and concomitant adoption of the MolSSI QCARCHIVE INFRASTRUCTURE project, makes the latest version of PSI4 well suited to distributed computation of large numbers of independent tasks. The project has fostered the development of independent software components that may be reused in other quantum chemistry programs.
Figures

References
-
- Parrish R. M., Burns L. A., Smith D. G. A., Simmonett A. C., DePrince A. E. III, Hohenstein E. G., Bozkaya U., Sokolov A. Y., Di Remigio R., Richard R. M., Gonthier J. F., James A. M., McAlexander H. R., Kumar A., Saitow M., Wang X., Pritchard B. P., Verma P., Schaefer H. F. III, Patkowski K., King R. A., Valeev E. F., Evangelista F. A., Turney J. M., Crawford T. D., and Sherrill C. D., J. Chem. Theory Comput. 13, 3185 (2017).10.1021/acs.jctc.7b00174 - DOI - PMC - PubMed
-
- Jeziorski B., Moszynski R., and Szalewicz K., Chem. Rev. 94, 1887 (1994).10.1021/cr00031a008 - DOI
-
- Szalewicz K., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2, 254 (2012).10.1002/wcms.86 - DOI
-
- Raghavachari K., Trucks G. W., Pople J. A., and Head-Gordon M., Chem. Phys. Lett. 157, 479 (1989).10.1016/s0009-2614(89)87395-6 - DOI
LinkOut - more resources
Full Text Sources