

Differential diagnosis of vacuolar myopathies in the NGS era
- PMID: 32419263
- PMCID: PMC8017999
- DOI: 10.1111/bpa.12864
Differential diagnosis of vacuolar myopathies in the NGS era
Abstract
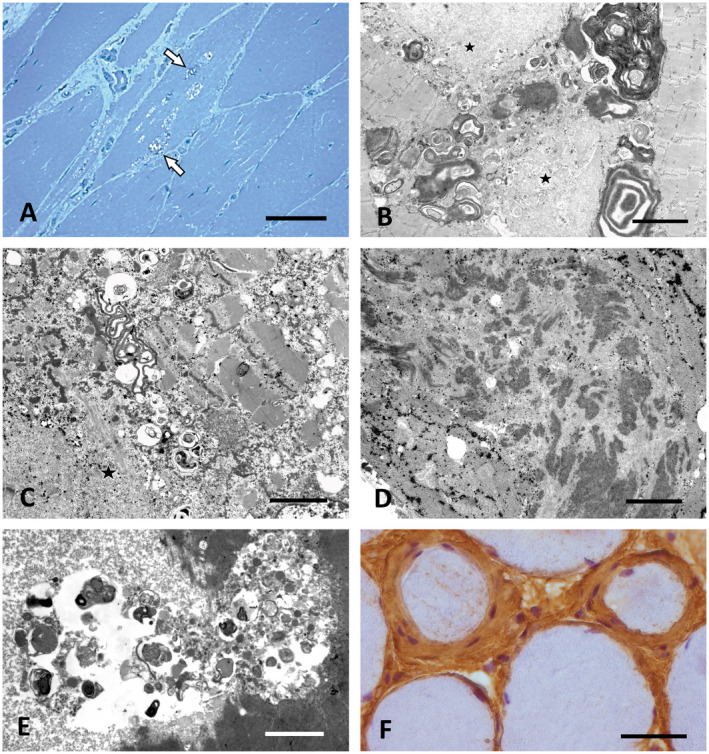
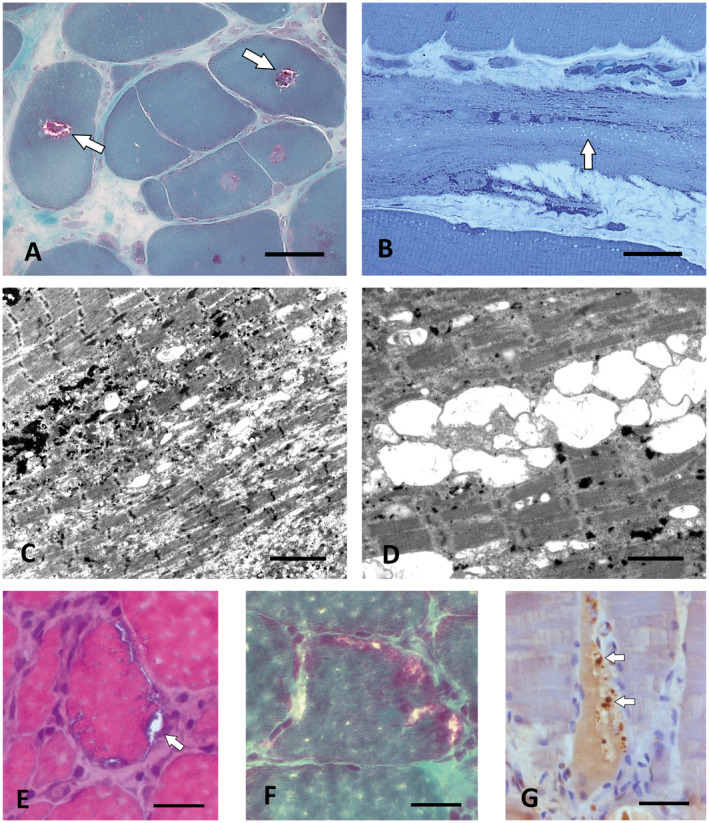
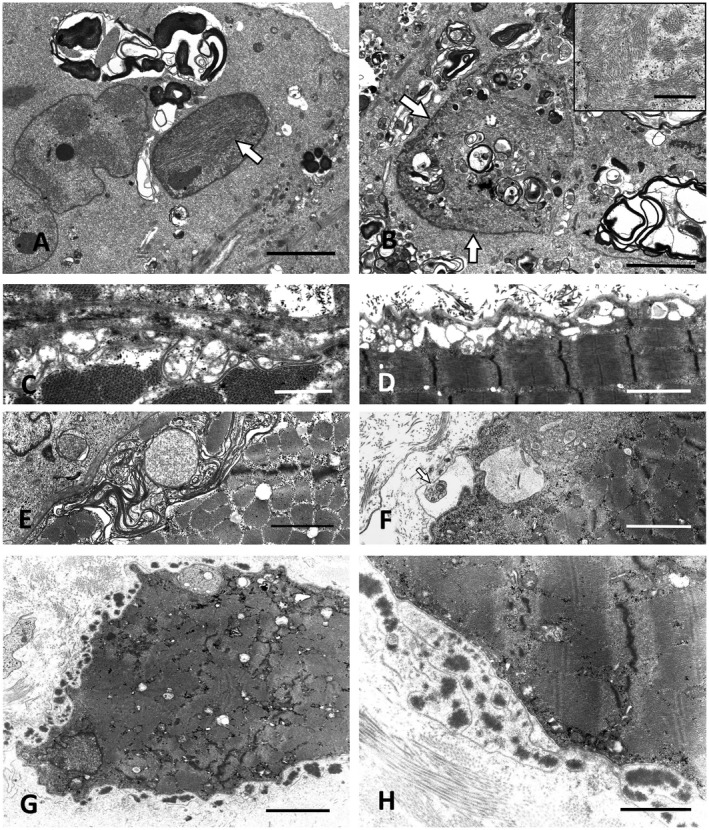
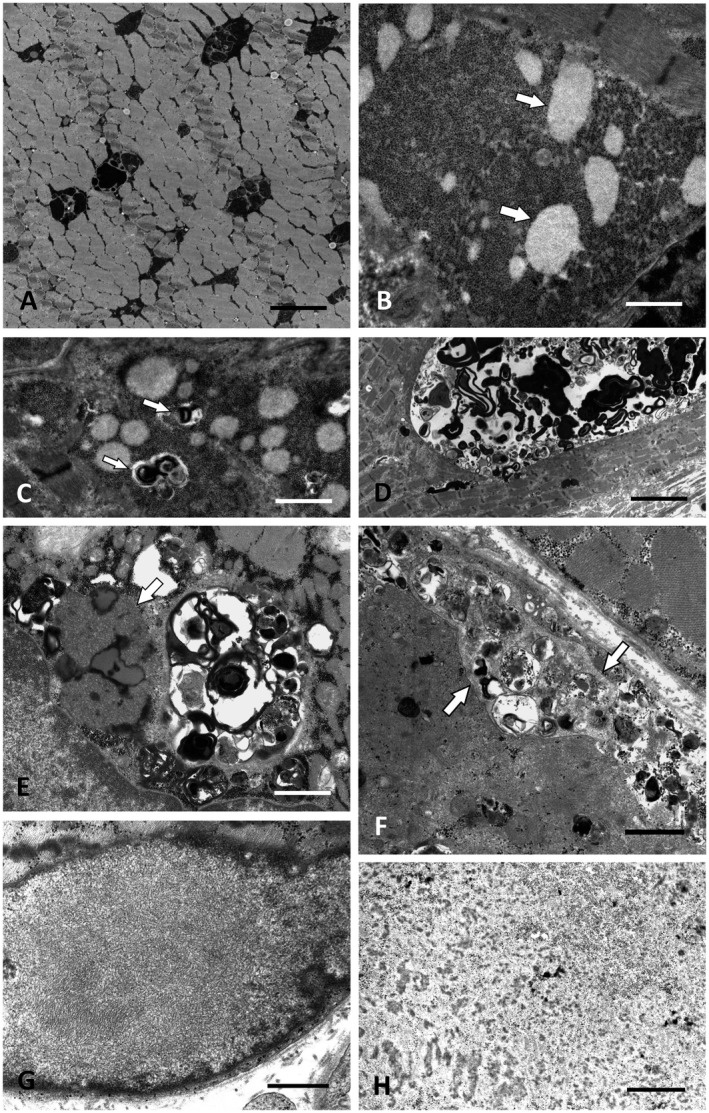
Altered autophagy accompanied by abnormal autophagic (rimmed) vacuoles detectable by light and electron microscopy is a common denominator of many familial and sporadic non-inflammatory muscle diseases. Even in the era of next generation sequencing (NGS), late-onset vacuolar myopathies remain a diagnostic challenge. We identified 32 adult vacuolar myopathy patients from 30 unrelated families, studied their clinical, histopathological and ultrastructural characteristics and performed genetic testing in index patients and relatives using Sanger sequencing and NGS including whole exome sequencing (WES). We established a molecular genetic diagnosis in 17 patients. Pathogenic mutations were found in genes typically linked to vacuolar myopathy (GNE, LDB3/ZASP, MYOT, DES and GAA), but also in genes not regularly associated with severely altered autophagy (FKRP, DYSF, CAV3, COL6A2, GYG1 and TRIM32) and in the digenic facioscapulohumeral muscular dystrophy 2. Characteristic histopathological features including distinct patterns of myofibrillar disarray and evidence of exocytosis proved to be helpful to distinguish causes of vacuolar myopathies. Biopsy validated the pathogenicity of the novel mutations p.(Phe55*) and p.(Arg216*) in GYG1 and of the p.(Leu156Pro) TRIM32 mutation combined with compound heterozygous deletion of exon 2 of TRIM32 and expanded the phenotype of Ala93Thr-caveolinopathy and of limb-girdle muscular dystrophy 2i caused by FKRP mutation. In 15 patients no causal variants were detected by Sanger sequencing and NGS panel analysis. In 12 of these cases, WES was performed, but did not yield any definite mutation or likely candidate gene. In one of these patients with a family history of muscle weakness, the vacuolar myopathy was eventually linked to chloroquine therapy. Our study illustrates the wide phenotypic and genotypic heterogeneity of vacuolar myopathies and validates the role of histopathology in assessing the pathogenicity of novel mutations detected by NGS. In a sizable portion of vacuolar myopathy cases, it remains to be shown whether the cause is hereditary or degenerative.
Keywords: FSHD; Pompe disease; TRIM32; autophagy; glycogenin 1; muscular dystrophy; myofibrillar myopathy; next generation sequencing (NGS); sarcotubular myopathy; vacuolar myopathy.
© 2020 The Authors. Brain Pathology published by John Wiley & Sons Ltd on behalf of International Society of Neuropathology.
Figures
References
-
- Atsumi T, Ishikawa S, Miyatake T, Yoshida M (1979) Myopathy and primary aldosteronism: electronmicroscopic study. Neurology 29:1348–1353. - PubMed
-
- Berardo A, Lornage X, Johari M, Evangelista T, Cejas C, Barroso F et al (2019) HNRNPDL‐related muscular dystrophy: expanding the clinical, morphological and MRI phenotypes. J Neurol 266:2524–2534. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
