

Humanization of yeast genes with multiple human orthologs reveals functional divergence between paralogs
- PMID: 32421706
- PMCID: PMC7259792
- DOI: 10.1371/journal.pbio.3000627
Humanization of yeast genes with multiple human orthologs reveals functional divergence between paralogs
Abstract
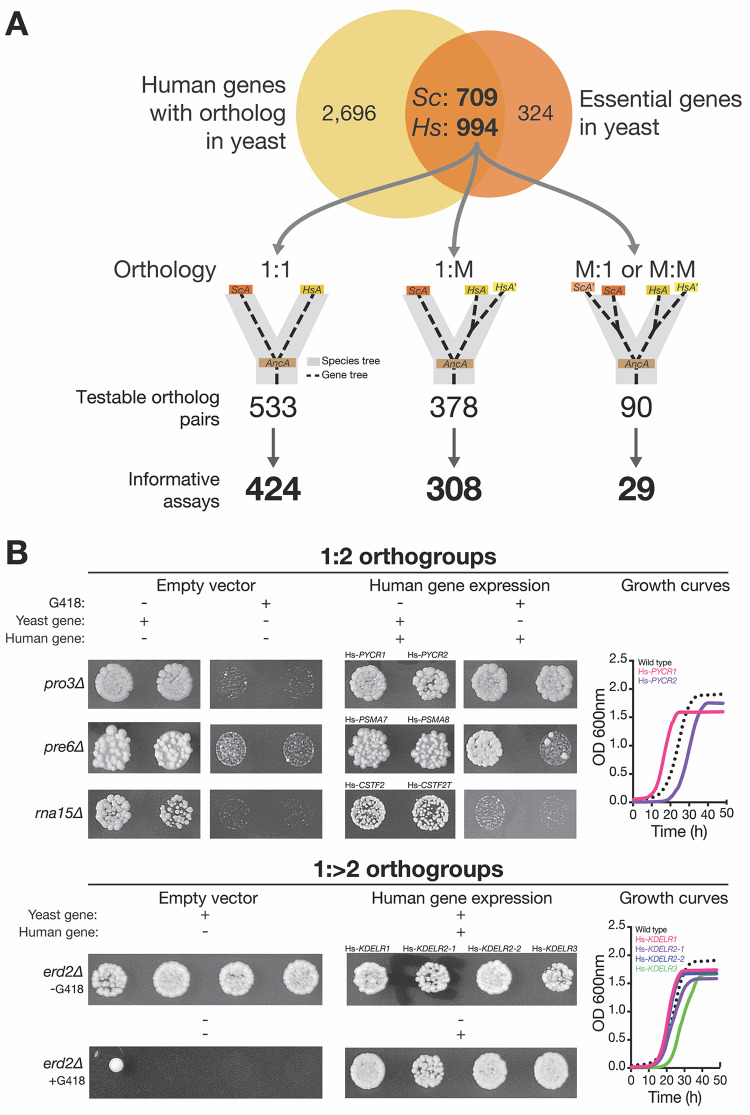
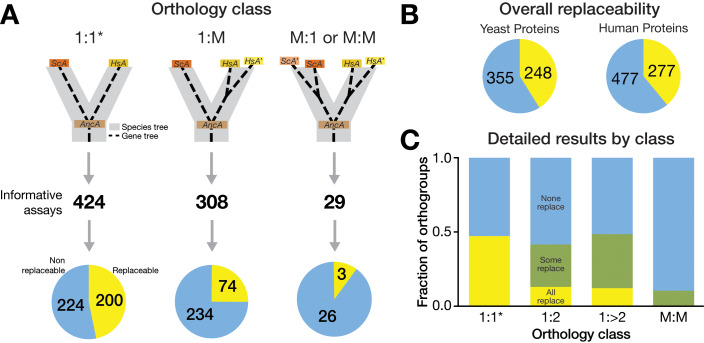
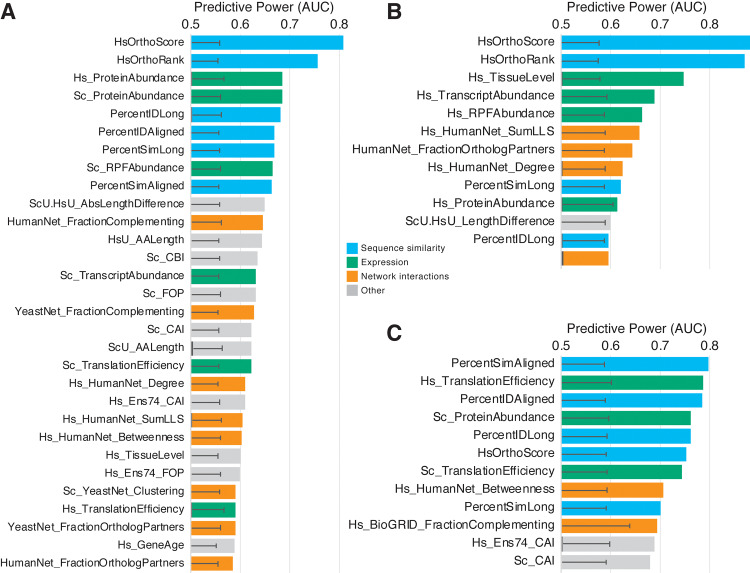
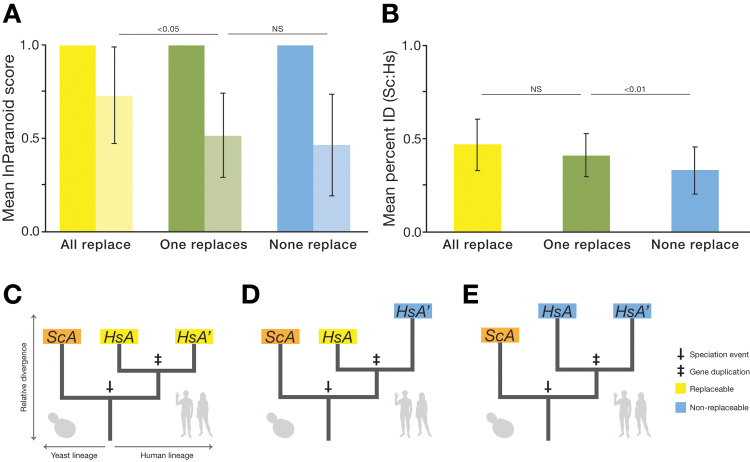
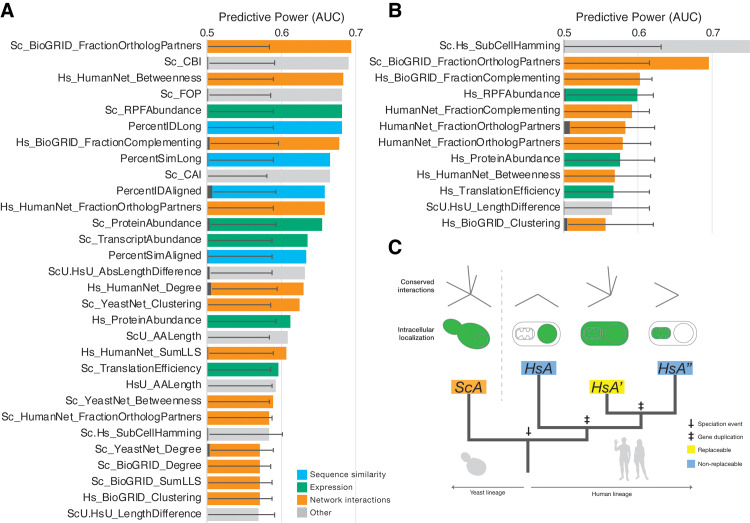
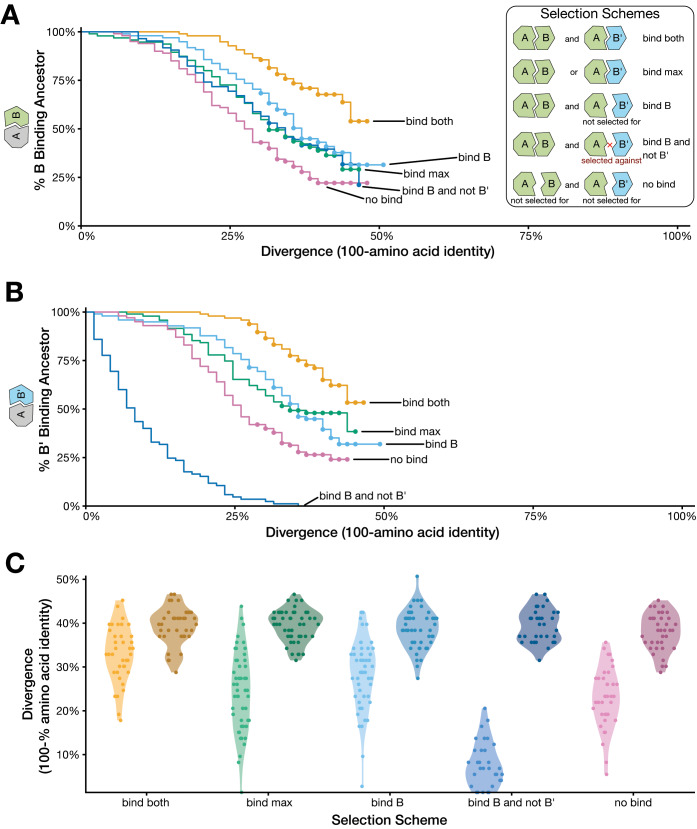
Despite over a billion years of evolutionary divergence, several thousand human genes possess clearly identifiable orthologs in yeast, and many have undergone lineage-specific duplications in one or both lineages. These duplicated genes may have been free to diverge in function since their expansion, and it is unclear how or at what rate ancestral functions are retained or partitioned among co-orthologs between species and within gene families. Thus, in order to investigate how ancestral functions are retained or lost post-duplication, we systematically replaced hundreds of essential yeast genes with their human orthologs from gene families that have undergone lineage-specific duplications, including those with single duplications (1 yeast gene to 2 human genes, 1:2) or higher-order expansions (1:>2) in the human lineage. We observe a variable pattern of replaceability across different ortholog classes, with an obvious trend toward differential replaceability inside gene families, and rarely observe replaceability by all members of a family. We quantify the ability of various properties of the orthologs to predict replaceability, showing that in the case of 1:2 orthologs, replaceability is predicted largely by the divergence and tissue-specific expression of the human co-orthologs, i.e., the human proteins that are less diverged from their yeast counterpart and more ubiquitously expressed across human tissues more often replace their single yeast ortholog. These trends were consistent with in silico simulations demonstrating that when only one ortholog can replace its corresponding yeast equivalent, it tends to be the least diverged of the pair. Replaceability of yeast genes having more than 2 human co-orthologs was marked by retention of orthologous interactions in functional or protein networks as well as by more ancestral subcellular localization. Overall, we performed >400 human gene replaceability assays, revealing 50 new human-yeast complementation pairs, thus opening up avenues to further functionally characterize these human genes in a simplified organismal context.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
