

Transcriptome-wide organization of subcellular microenvironments revealed by ATLAS-Seq
- PMID: 32421779
- PMCID: PMC7293051
- DOI: 10.1093/nar/gkaa334
Transcriptome-wide organization of subcellular microenvironments revealed by ATLAS-Seq
Abstract
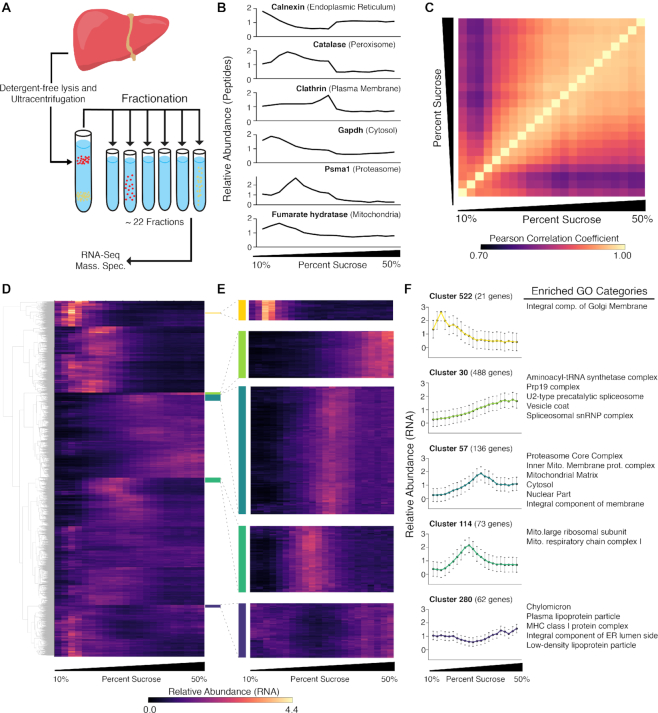
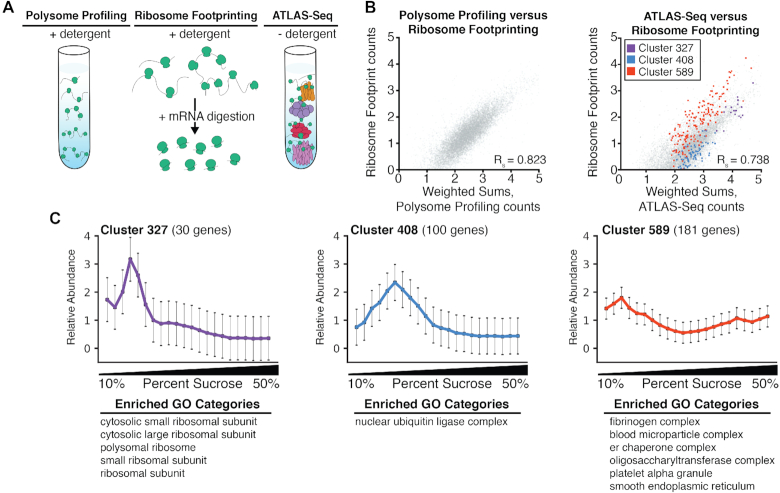
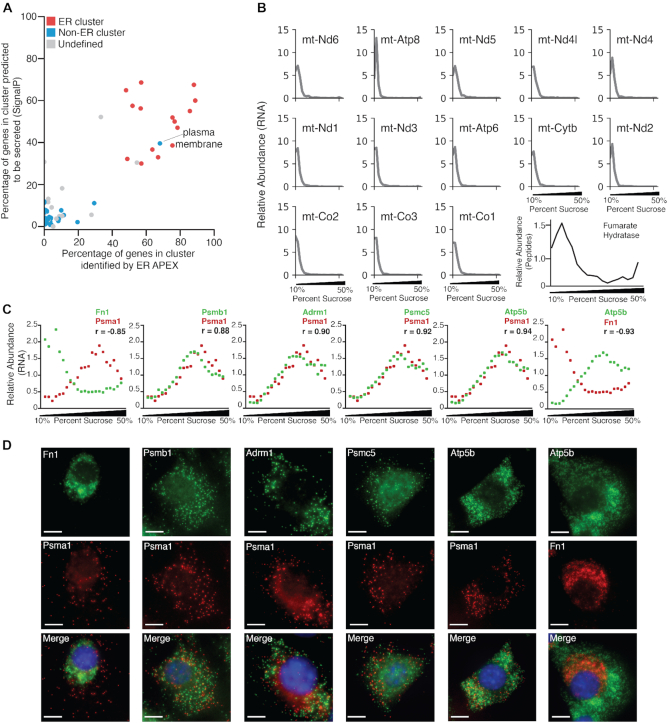
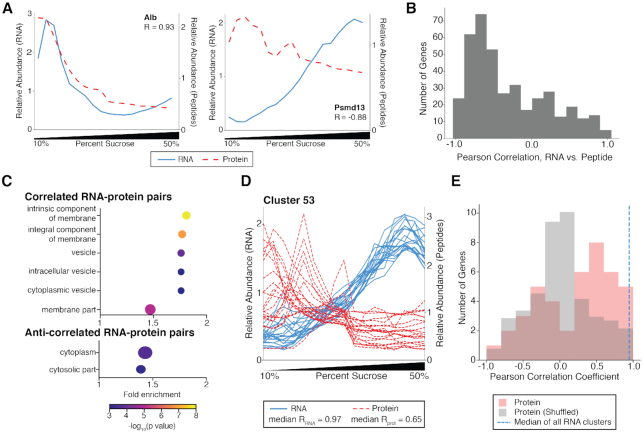
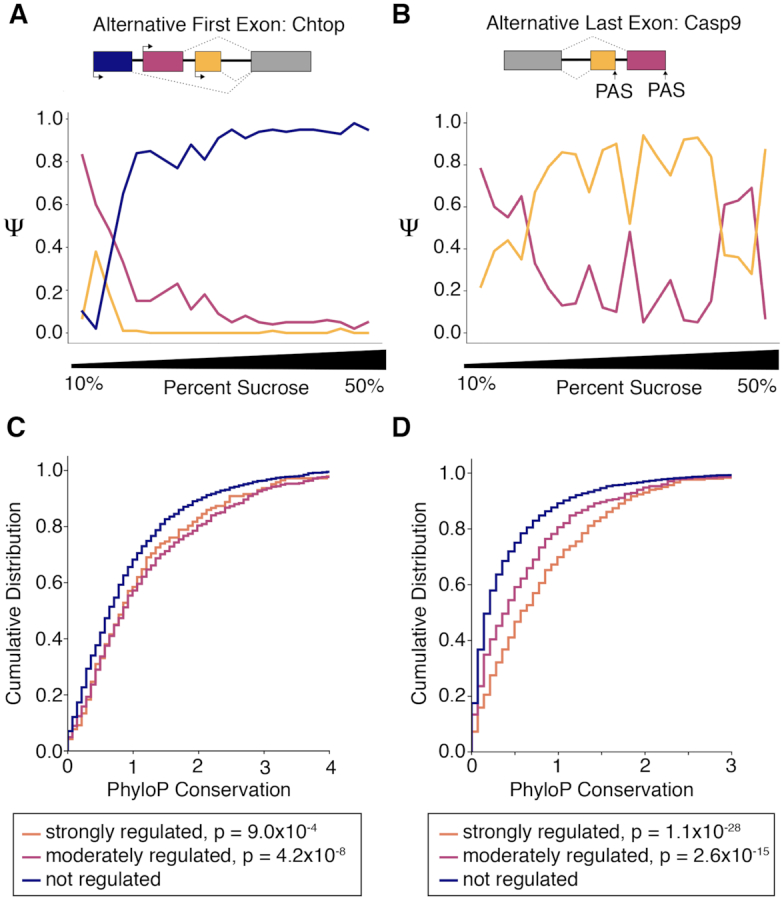
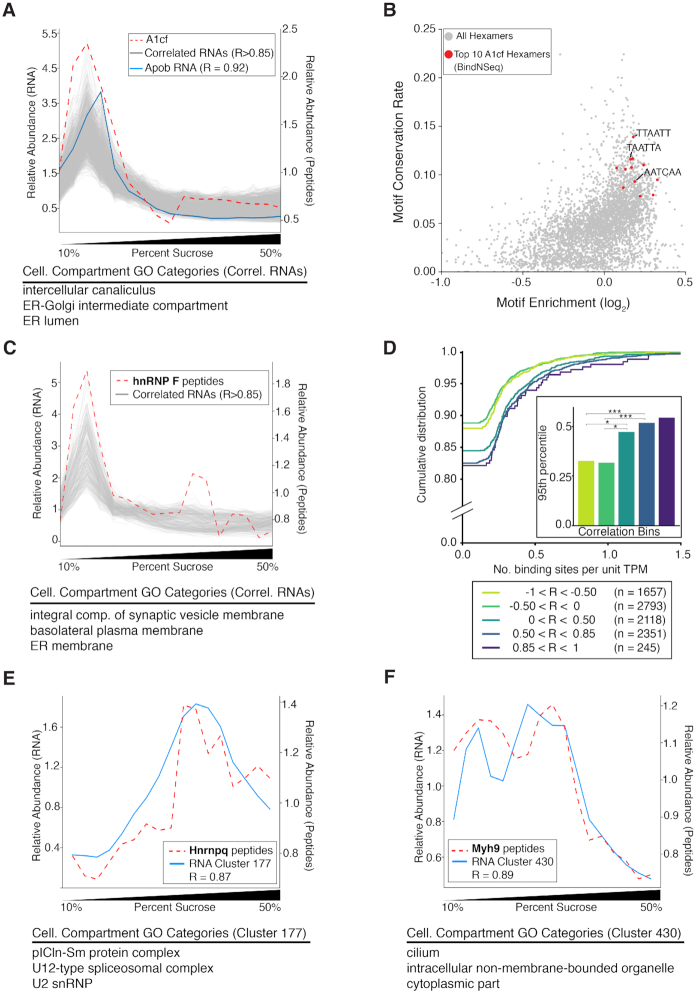
Subcellular organization of RNAs and proteins is critical for cell function, but we still lack global maps and conceptual frameworks for how these molecules are localized in cells and tissues. Here, we introduce ATLAS-Seq, which generates transcriptomes and proteomes from detergent-free tissue lysates fractionated across a sucrose gradient. Proteomic analysis of fractions confirmed separation of subcellular compartments. Unexpectedly, RNAs tended to co-sediment with other RNAs in similar protein complexes, cellular compartments, or with similar biological functions. With the exception of those encoding secreted proteins, most RNAs sedimented differently than their encoded protein counterparts. To identify RNA binding proteins potentially driving these patterns, we correlated their sedimentation profiles to all RNAs, confirming known interactions and predicting new associations. Hundreds of alternative RNA isoforms exhibited distinct sedimentation patterns across the gradient, despite sharing most of their coding sequence. These observations suggest that transcriptomes can be organized into networks of co-segregating mRNAs encoding functionally related proteins and provide insights into the establishment and maintenance of subcellular organization.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Steward O., Schuman E.M.. Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 2001; 24:299–325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
