

The proteasome activator REGγ accelerates cardiac hypertrophy by declining PP2Acα-SOD2 pathway
- PMID: 32424140
- PMCID: PMC7494903
- DOI: 10.1038/s41418-020-0554-8
The proteasome activator REGγ accelerates cardiac hypertrophy by declining PP2Acα-SOD2 pathway
Abstract
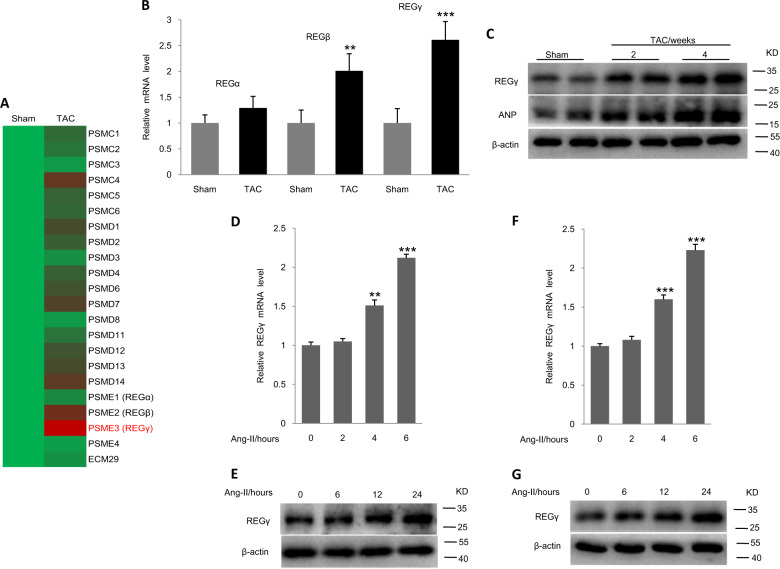
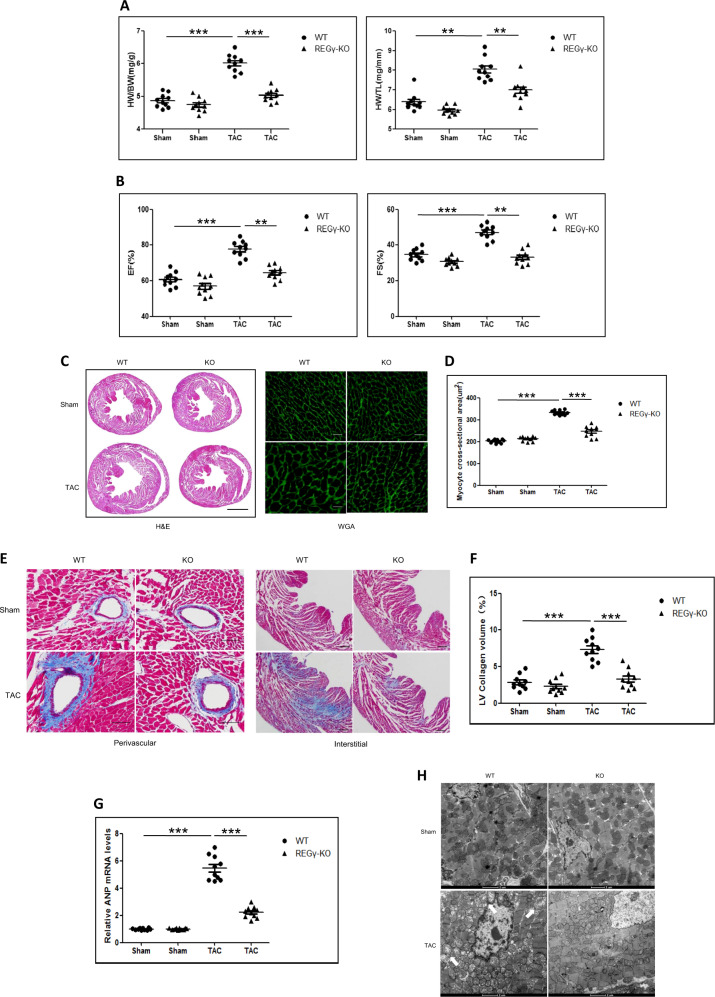
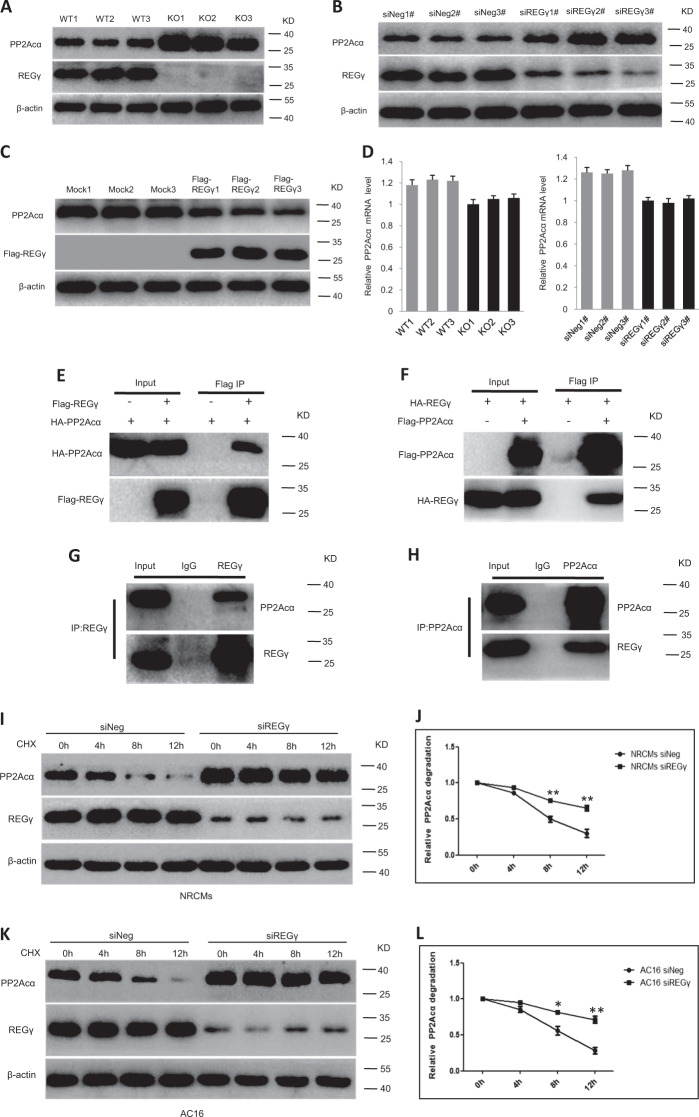
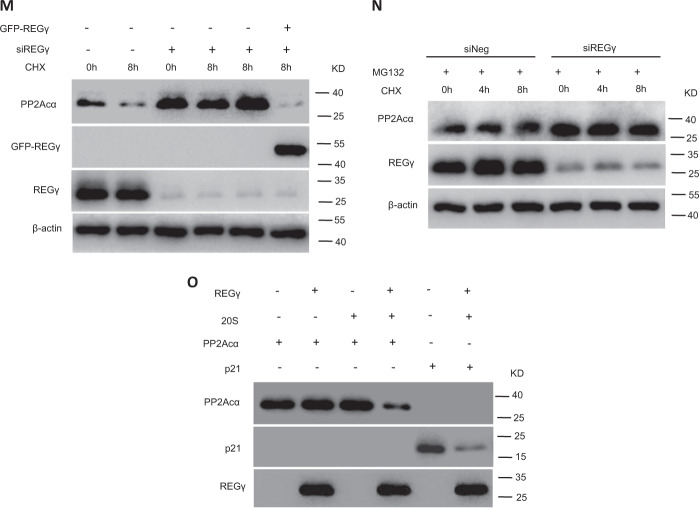
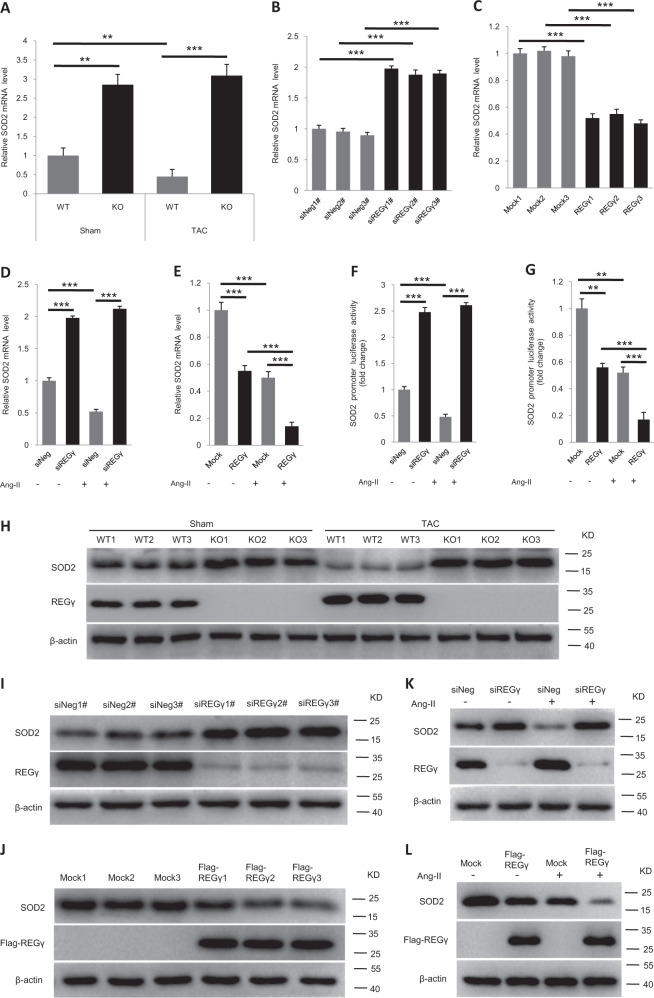
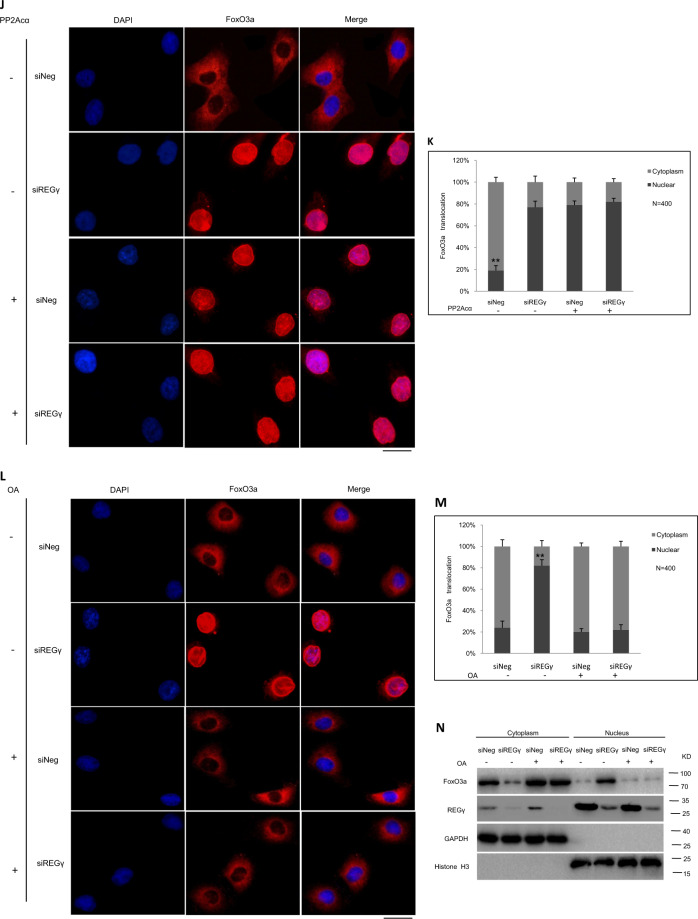
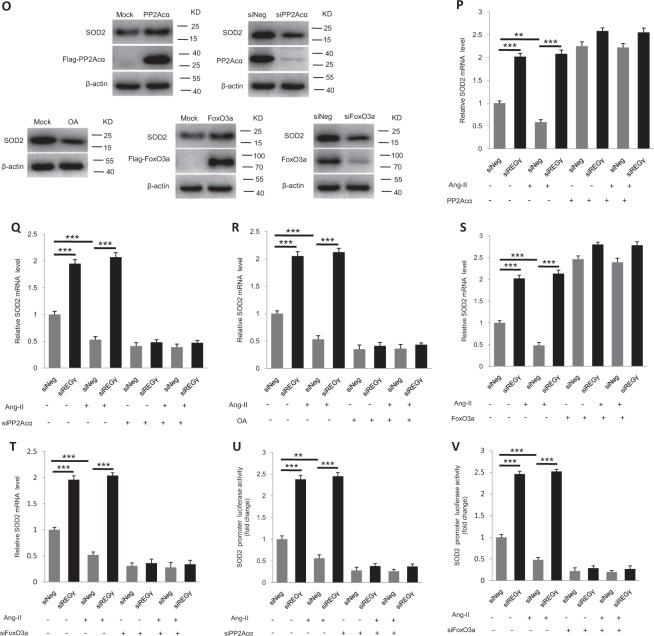
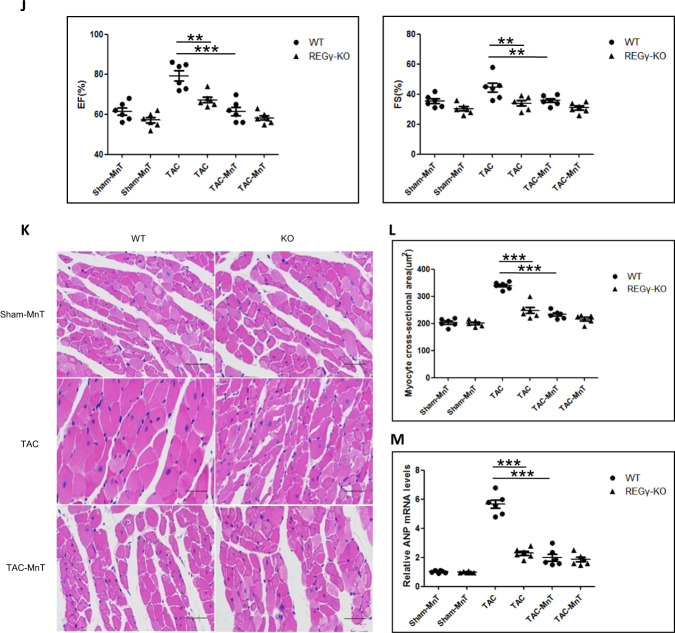
Pathological cardiac hypertrophy eventually leads to heart failure without adequate treatment. REGγ is emerging as 11S proteasome activator of 20S proteasome to promote the degradation of cellular proteins in a ubiquitin- and ATP-independent manner. Here, we found that REGγ was significantly upregulated in the transverse aortic constriction (TAC)-induced hypertrophic hearts and angiotensin II (Ang II)-treated cardiomyocytes. REGγ deficiency ameliorated pressure overload-induced cardiac hypertrophy were associated with inhibition of cardiac reactive oxygen species (ROS) accumulation and suppression of protein phosphatase 2A catalytic subunit α (PP2Acα) decay. Mechanistically, REGγ interacted with and targeted PP2Acα for degradation directly, thereby leading to increase of phosphorylation levels and nuclear export of Forkhead box protein O (FoxO) 3a and subsequent of SOD2 decline, ROS accumulation, and cardiac hypertrophy. Introducing exogenous PP2Acα or SOD2 to human cardiomyocytes significantly rescued the REGγ-mediated ROS accumulation of Ang II stimulation in vitro. Furthermore, treatment with superoxide dismutase mimetic, MnTBAP prevented cardiac ROS production and hypertrophy features that REGγ caused in vivo, thereby establishing a REGγ-PP2Acα-FoxO3a-SOD2 pathway in cardiac oxidative stress and hypertrophy, indicates modulating the REGγ-proteasome activity may be a potential therapeutic approach in cardiac hypertrophy-associated disorders.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures




References
-
- Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. - PubMed
-
- Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–8. - PubMed
-
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. - PubMed
-
- Varshavsky A. Regulated protein degradation. Trends Biochem Sci. 2005;30::283–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
