Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma
- PMID: 32424208
- PMCID: PMC7235245
- DOI: 10.1038/s41467-020-16295-5
Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma
Abstract
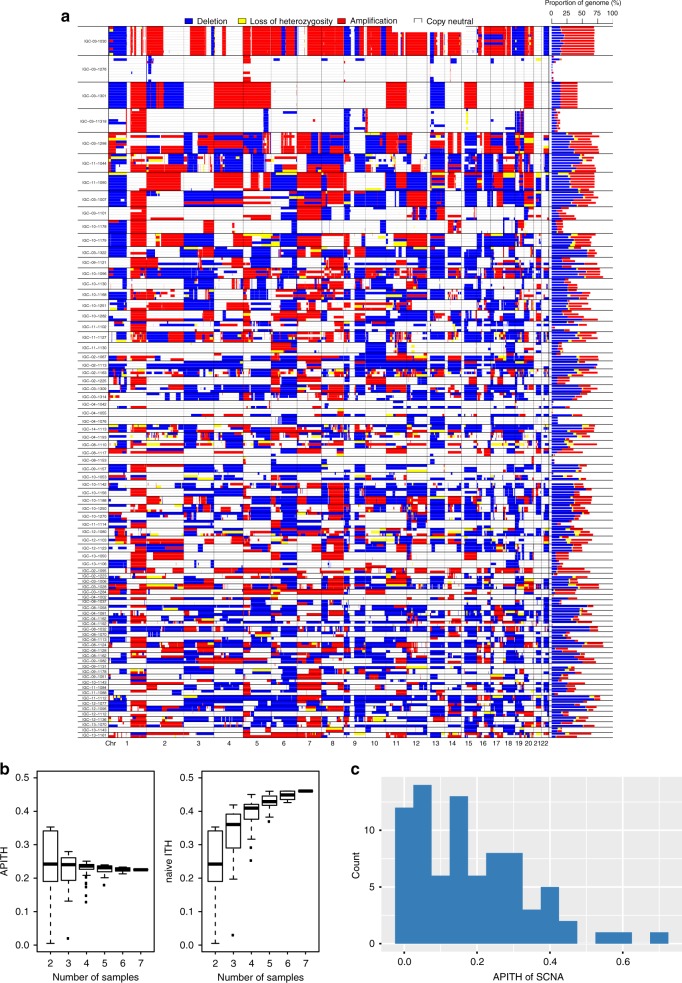
Intratumor heterogeneity (ITH) of genomic alterations may impact prognosis of lung adenocarcinoma (LUAD). Here, we investigate ITH of somatic copy number alterations (SCNAs), DNA methylation, and point mutations in lung cancer driver genes in 292 tumor samples from 84 patients with LUAD. LUAD samples show substantial SCNA and methylation ITH, and clonal architecture analyses present congruent evolutionary trajectories for SCNAs and DNA methylation aberrations. Methylation ITH mapping to gene promoter areas or tumor suppressor genes is low. Moreover, ITH composed of genetic and epigenetic mechanisms altering the same cancer driver genes is shown in several tumors. To quantify ITH for valid statistical association analyses, we develope an average pairwise ITH index (APITH), which does not depend on the number of samples per tumor. Both APITH indexes for SCNAs and methylation aberrations show significant associations with poor prognosis. This study further establishes the important clinical implications of genetic and epigenetic ITH in LUAD.
Conflict of interest statement
The authors declare no competing interests.
Figures