

Enhancer reprogramming driven by high-order assemblies of transcription factors promotes phenotypic plasticity and breast cancer endocrine resistance
- PMID: 32424275
- PMCID: PMC7737911
- DOI: 10.1038/s41556-020-0514-z
Enhancer reprogramming driven by high-order assemblies of transcription factors promotes phenotypic plasticity and breast cancer endocrine resistance
Abstract
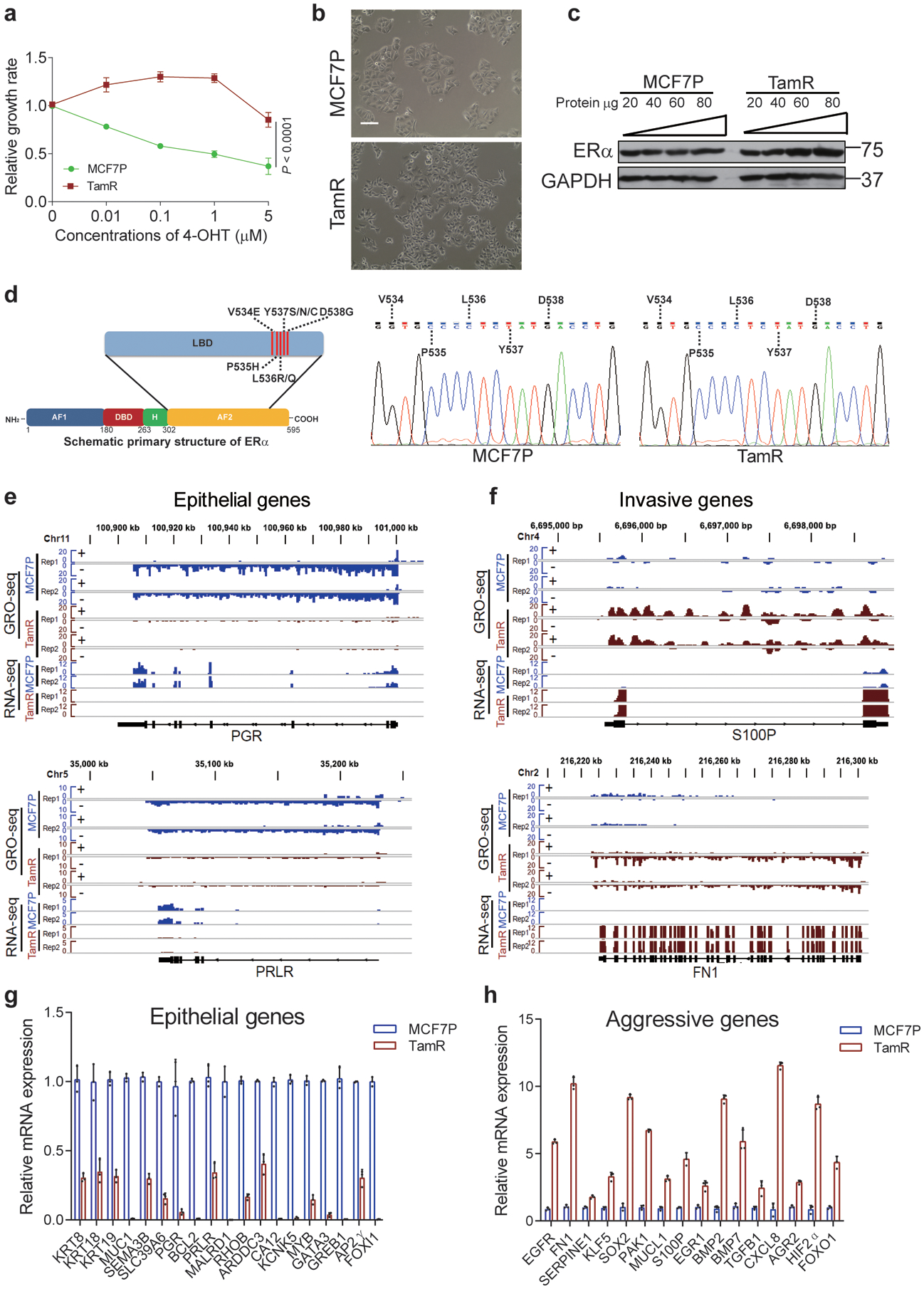
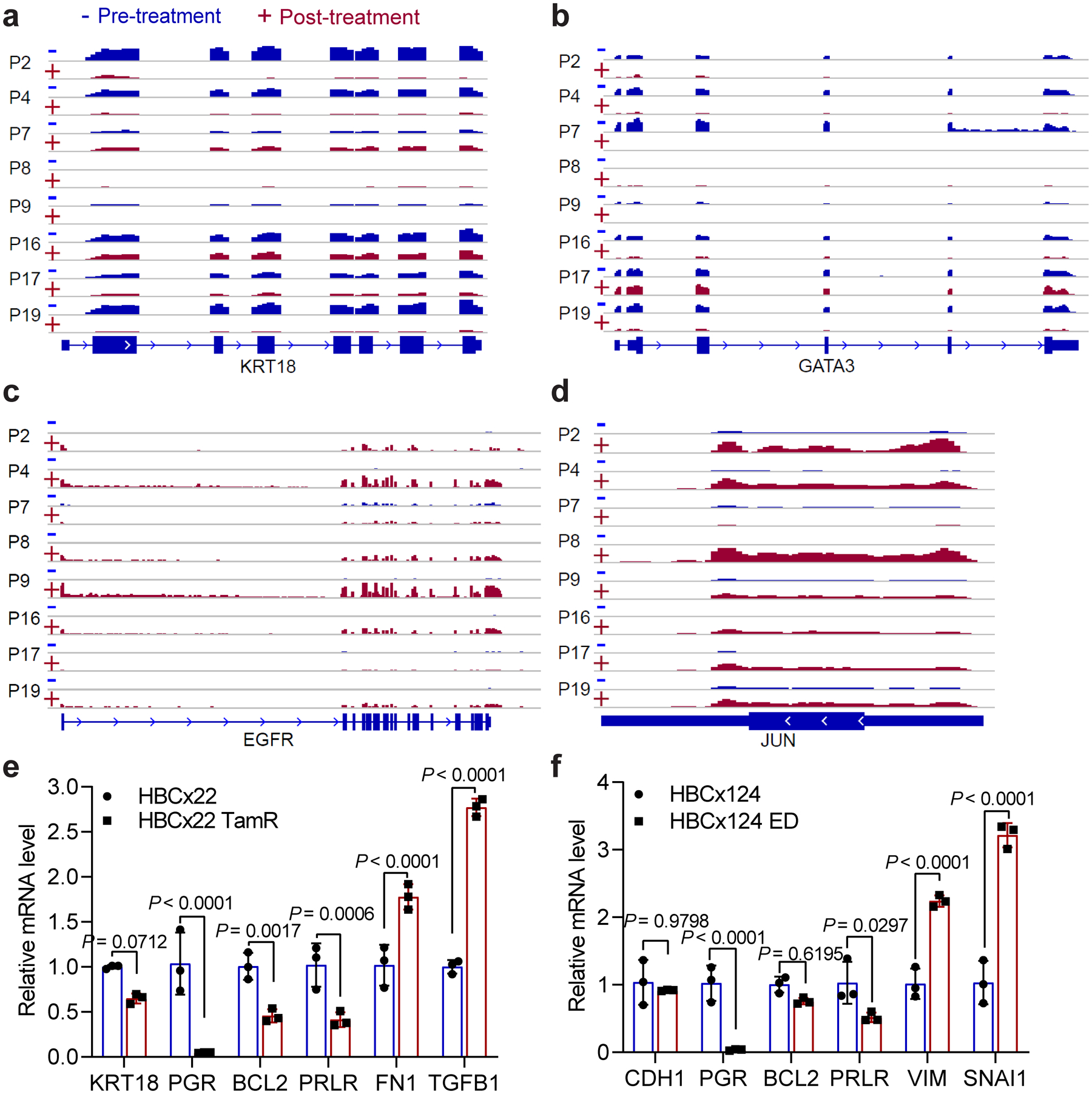
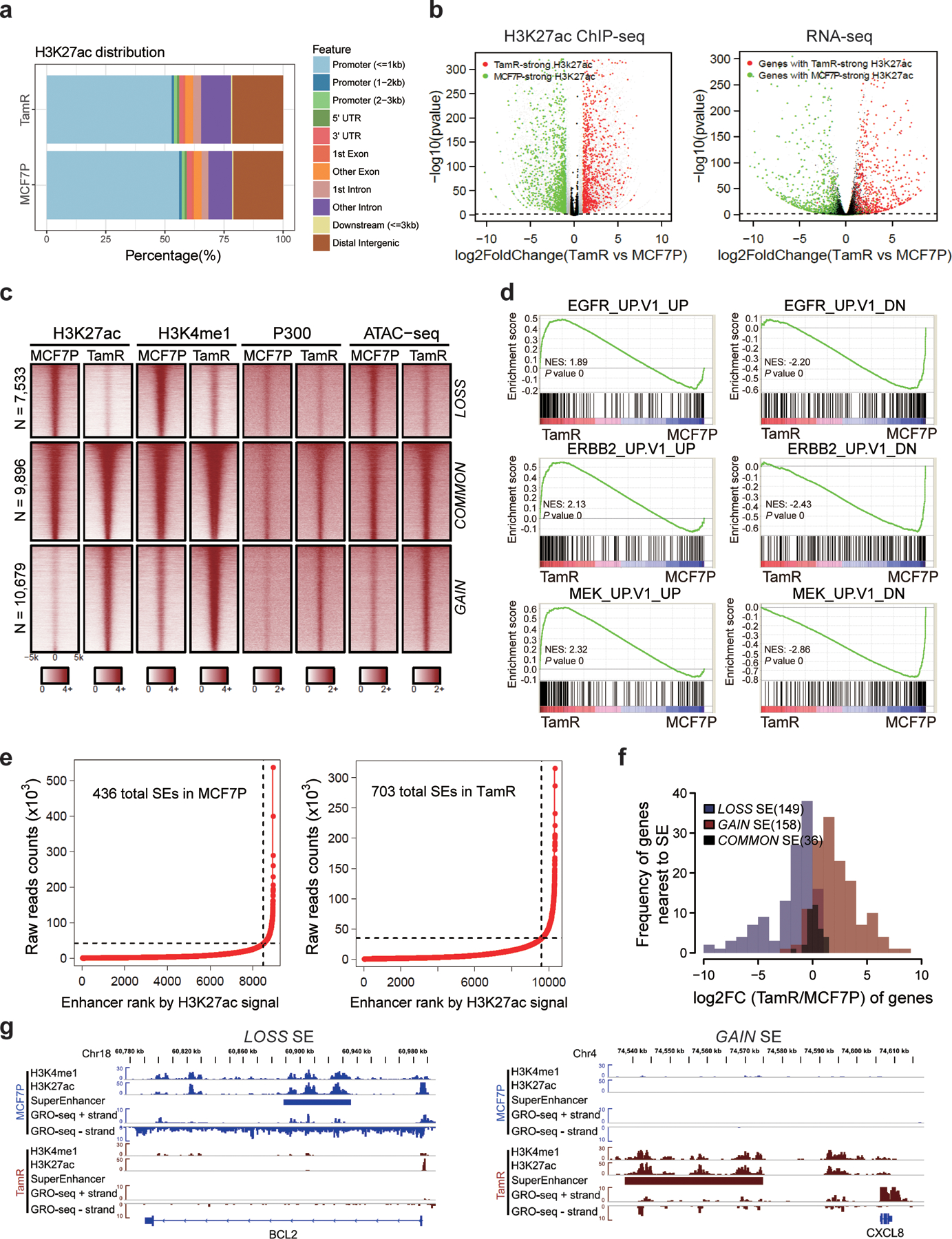
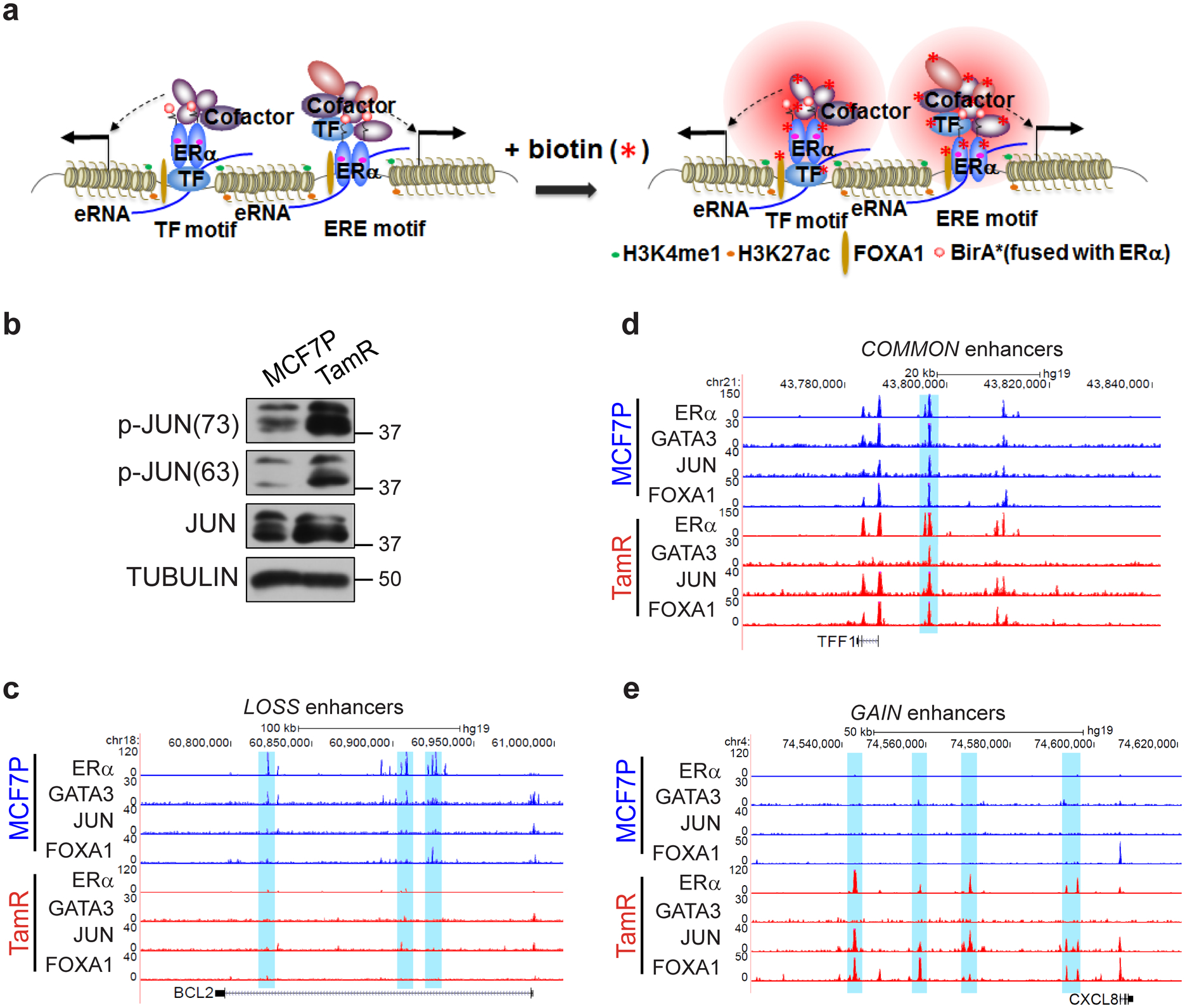
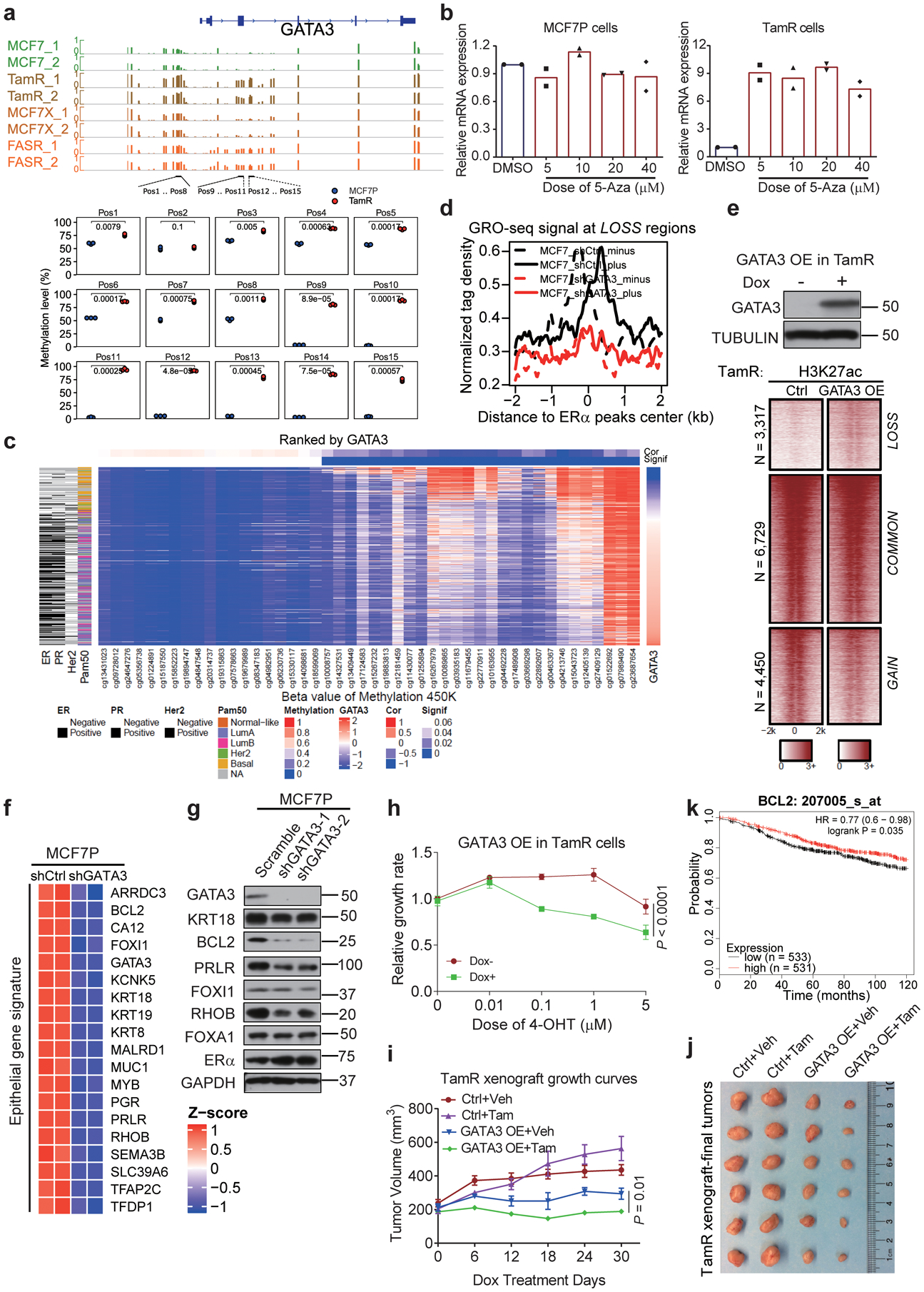
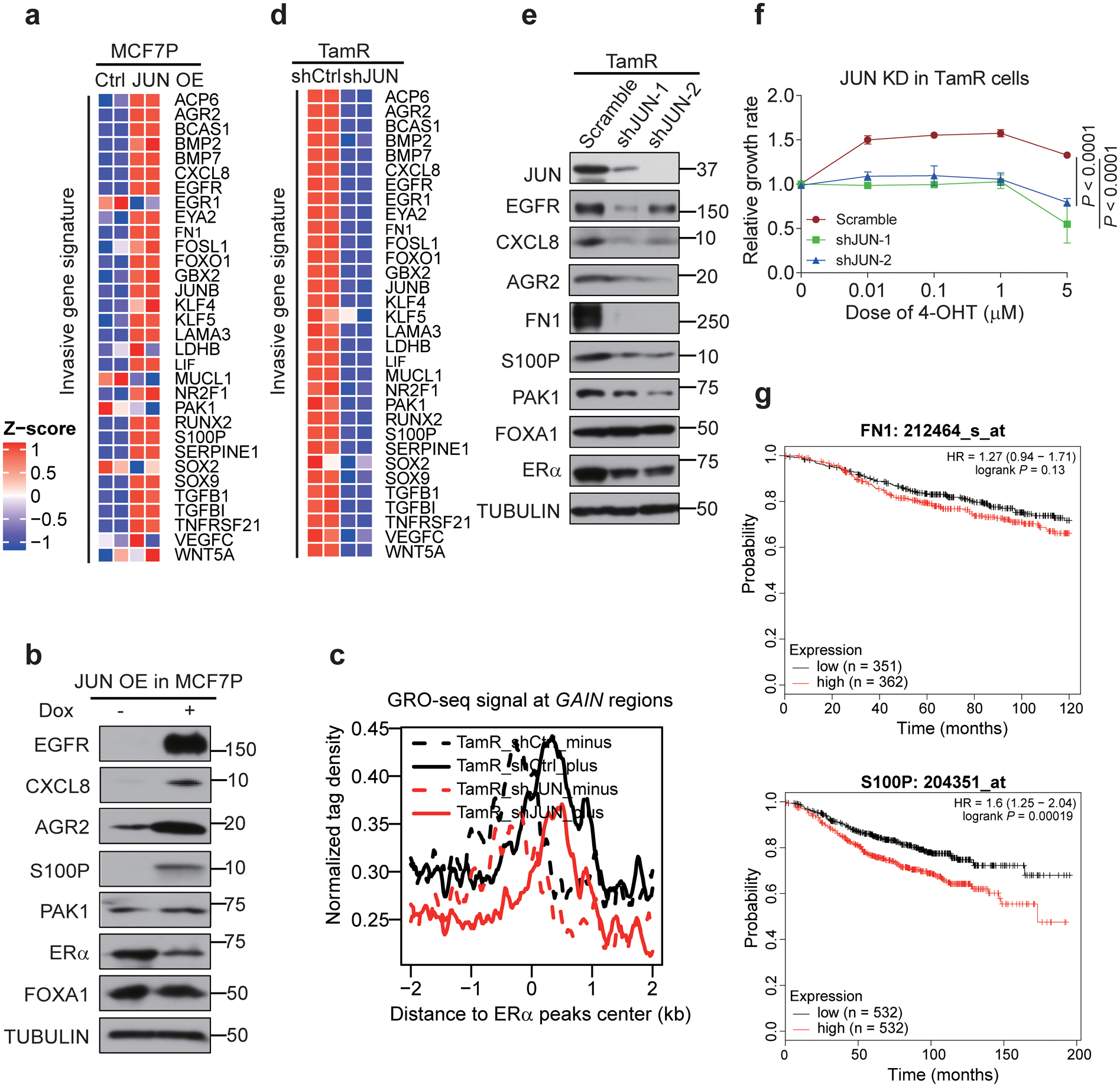
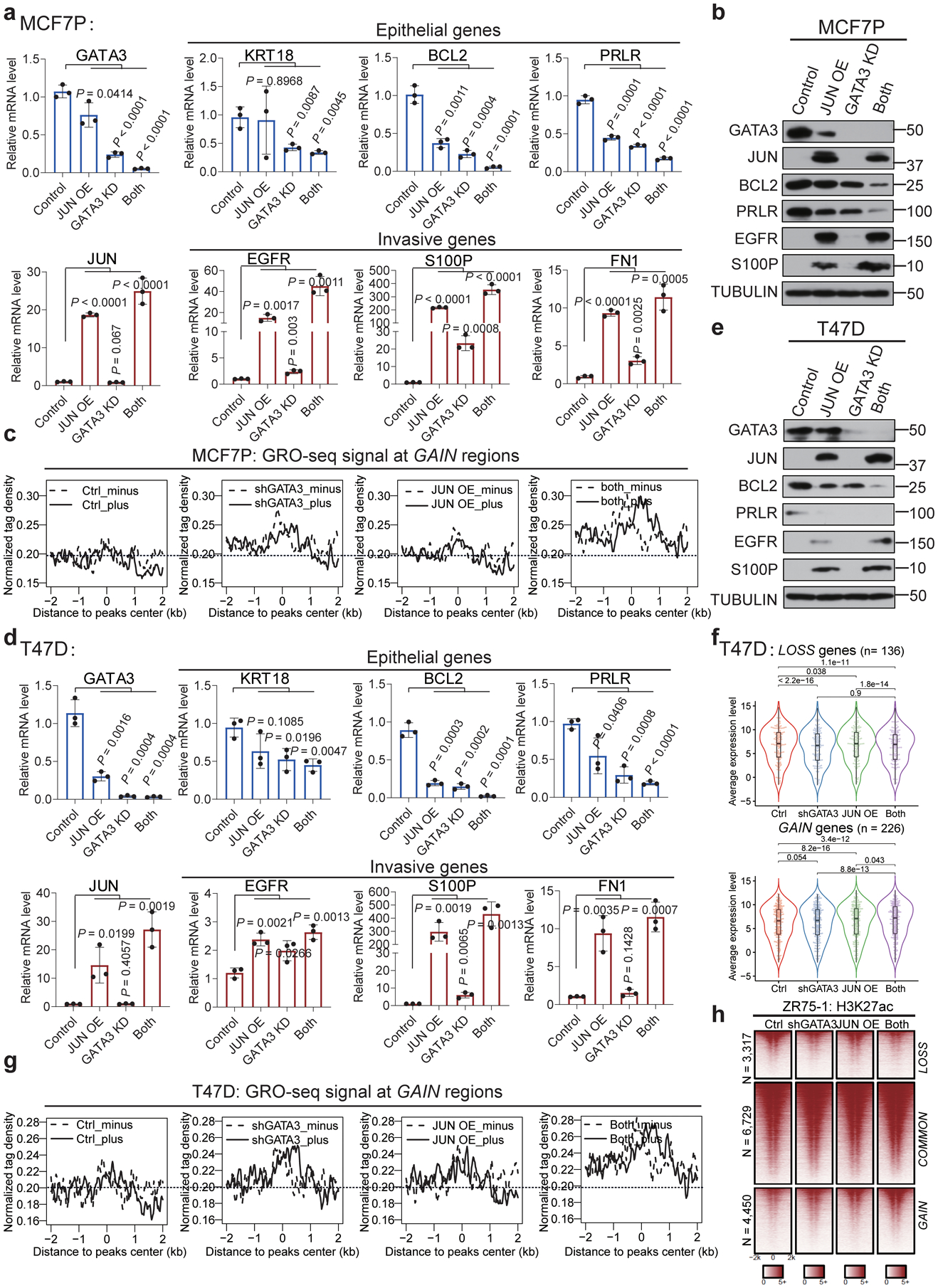
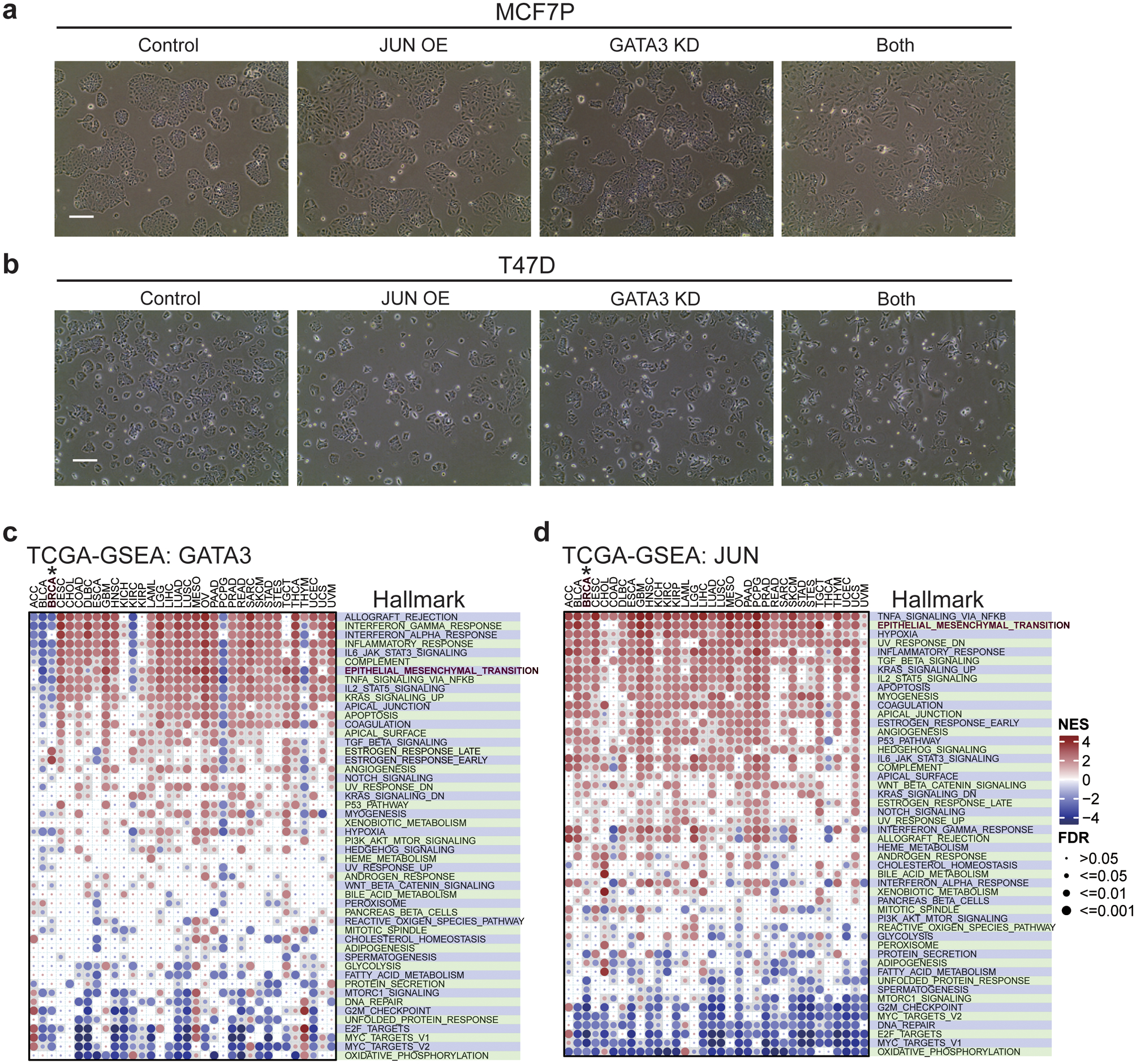
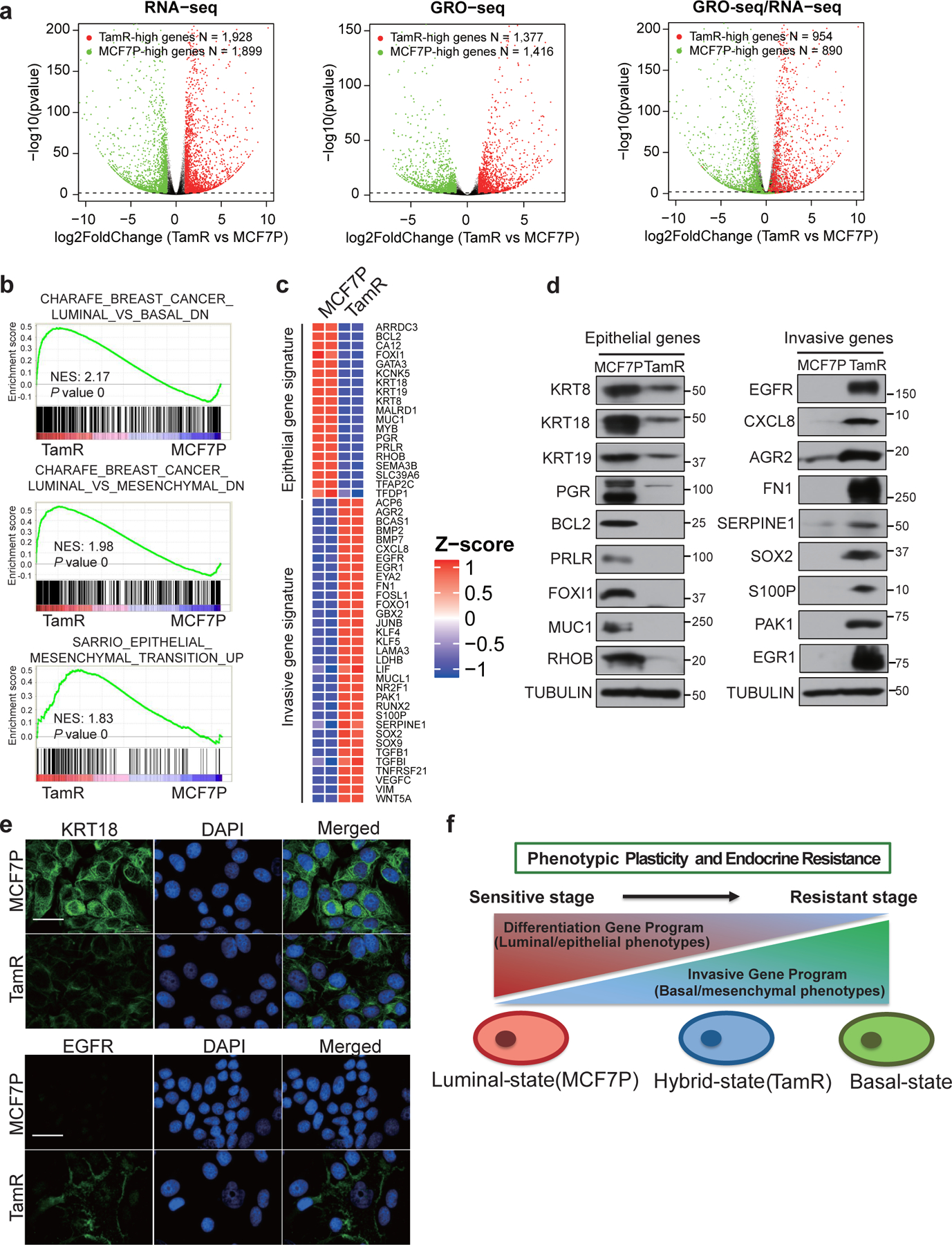
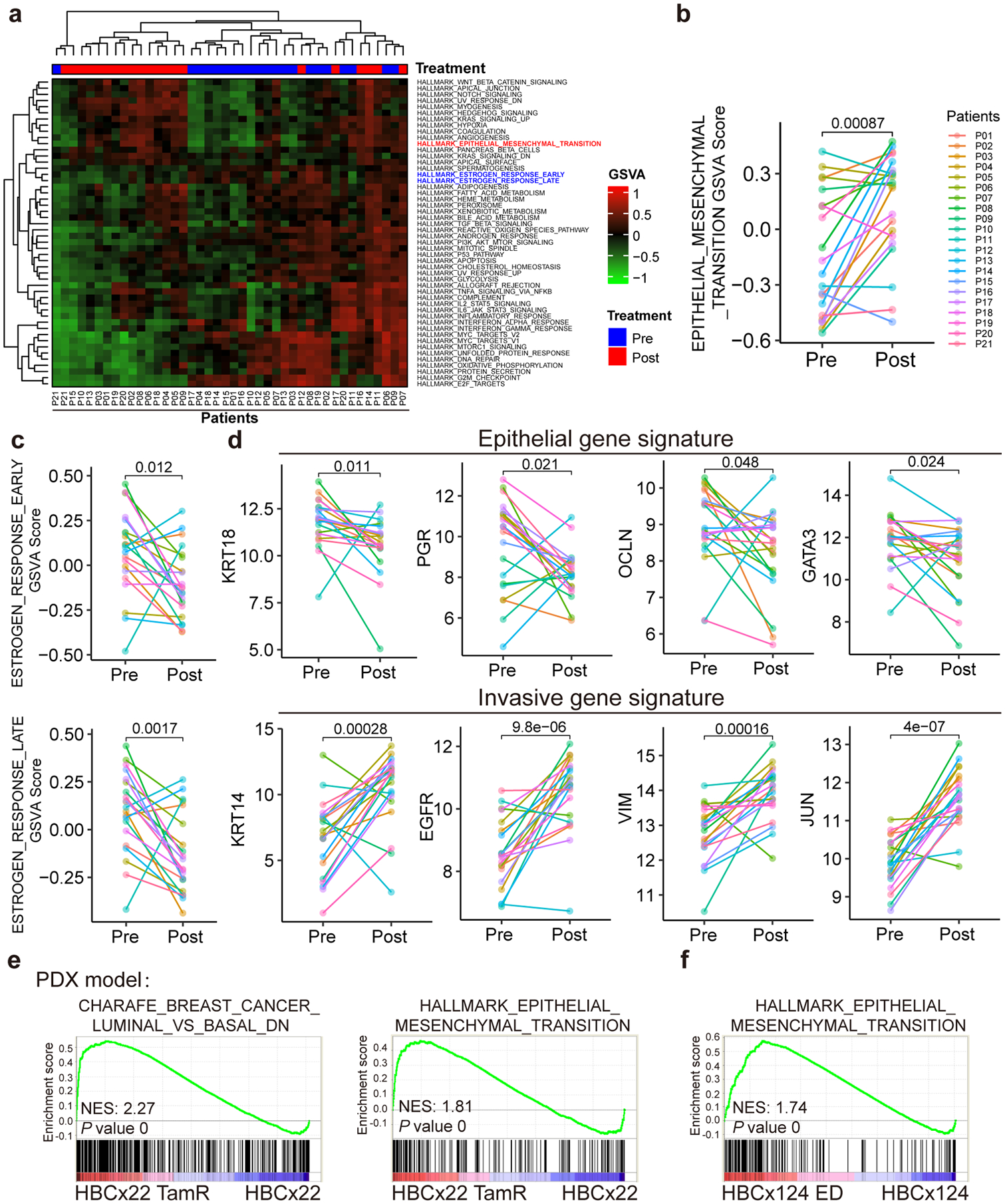
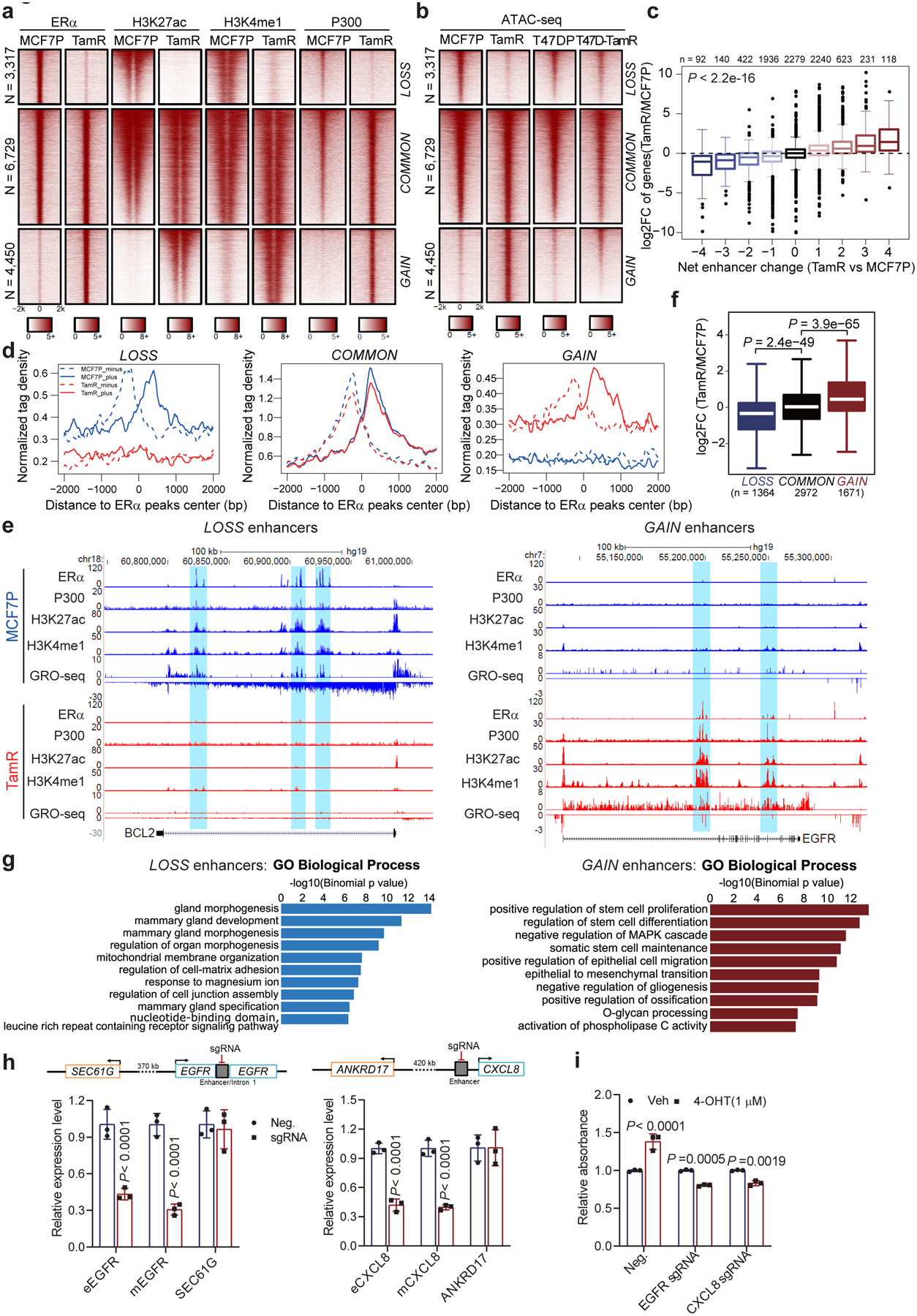
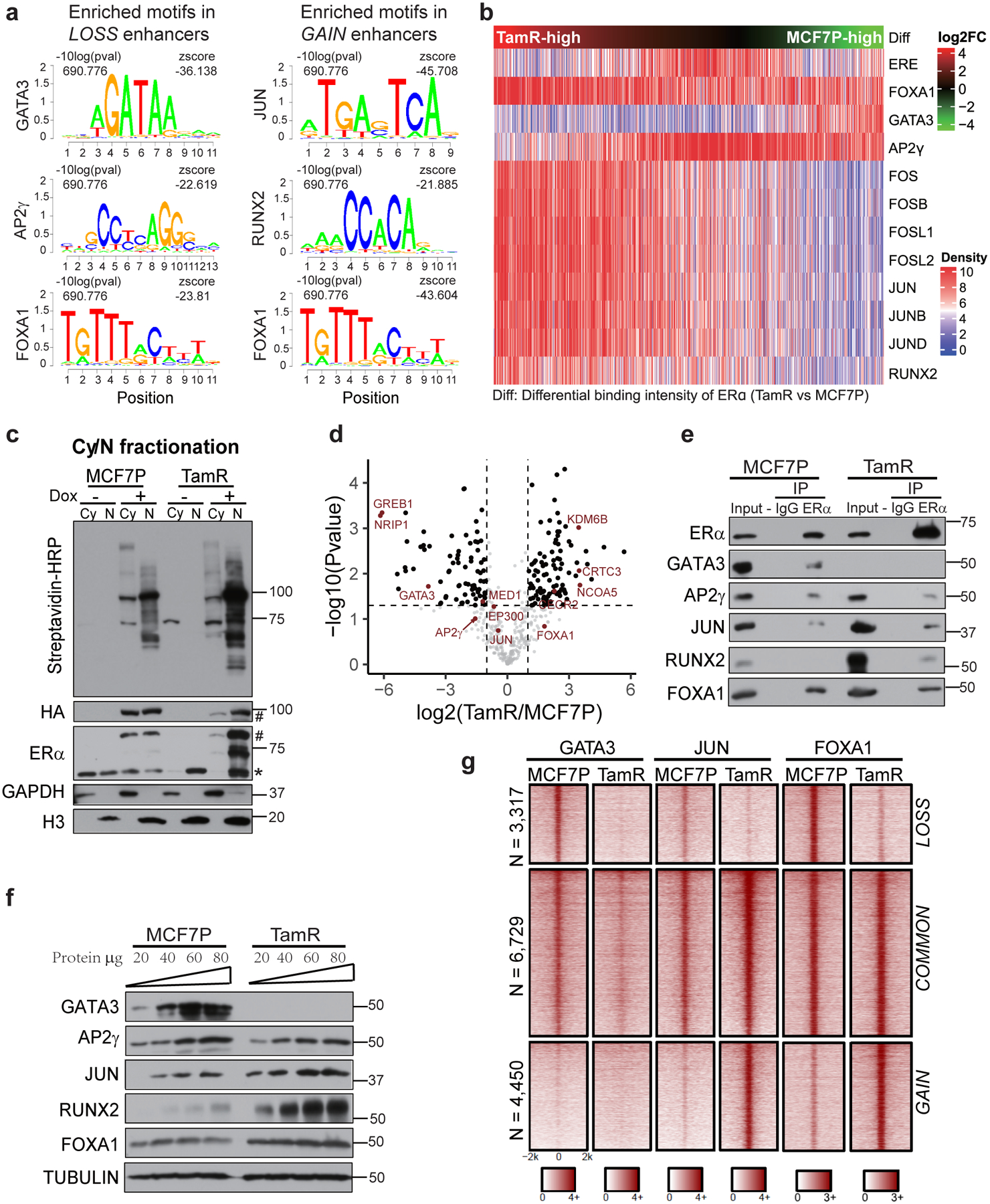
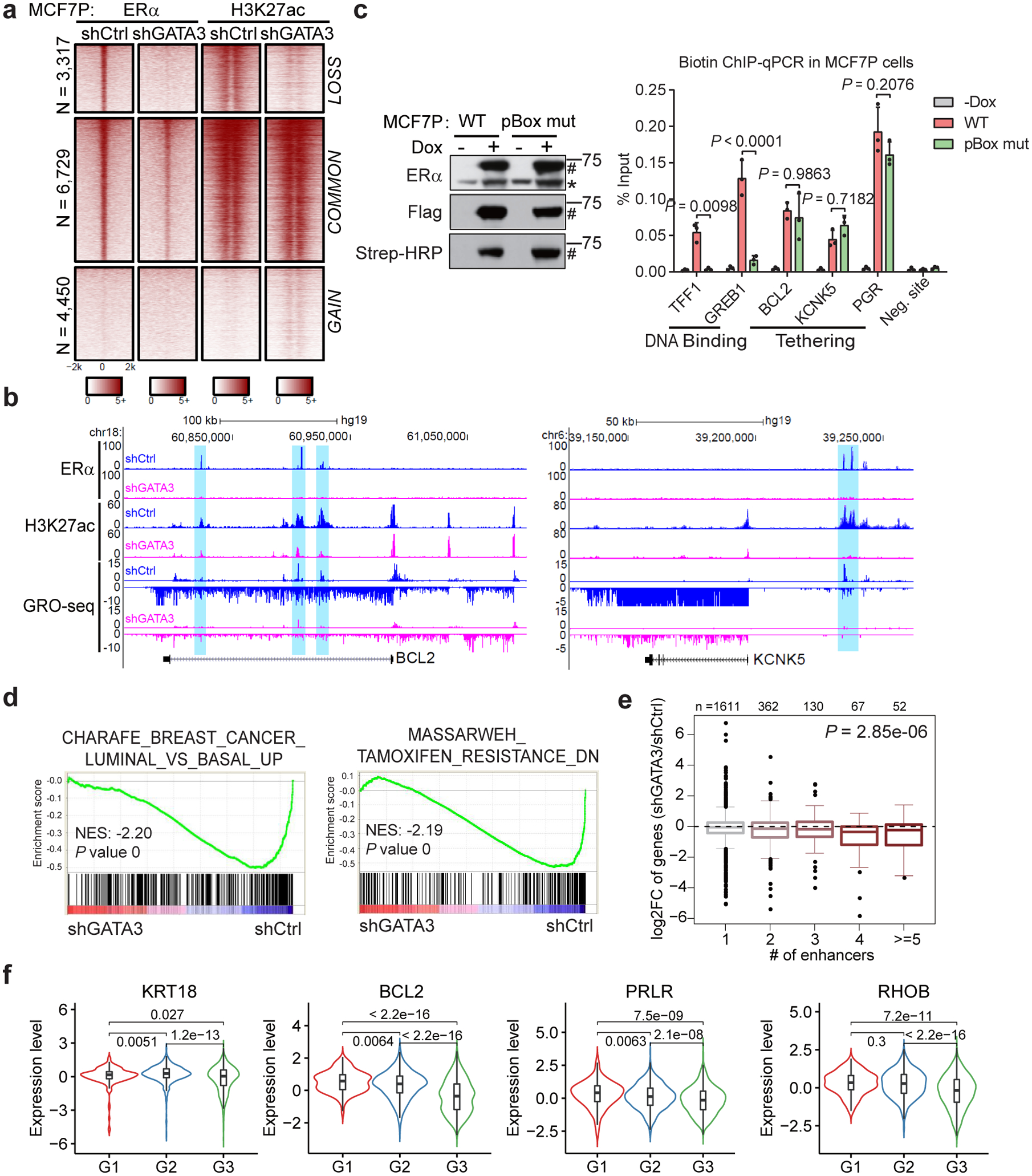
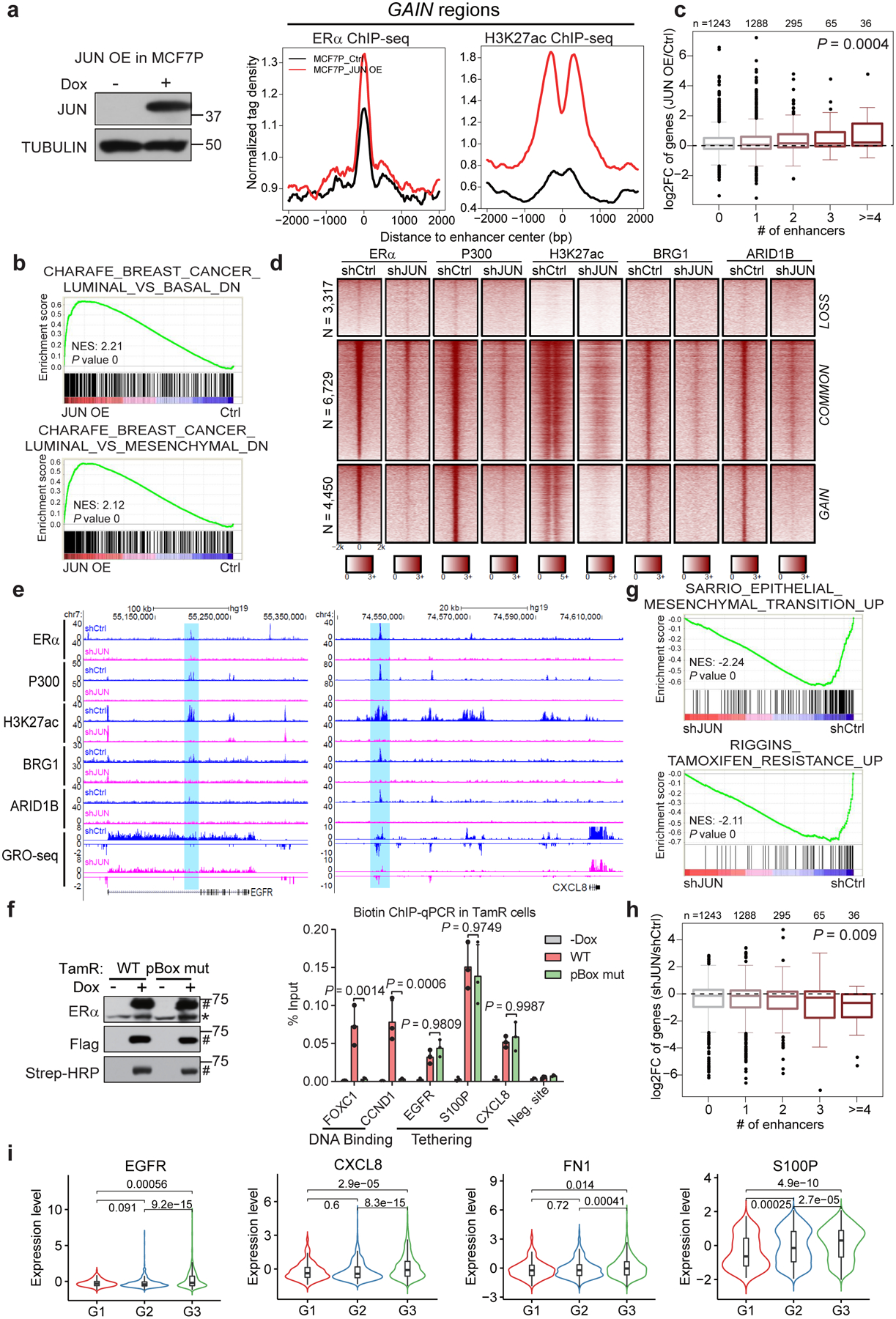
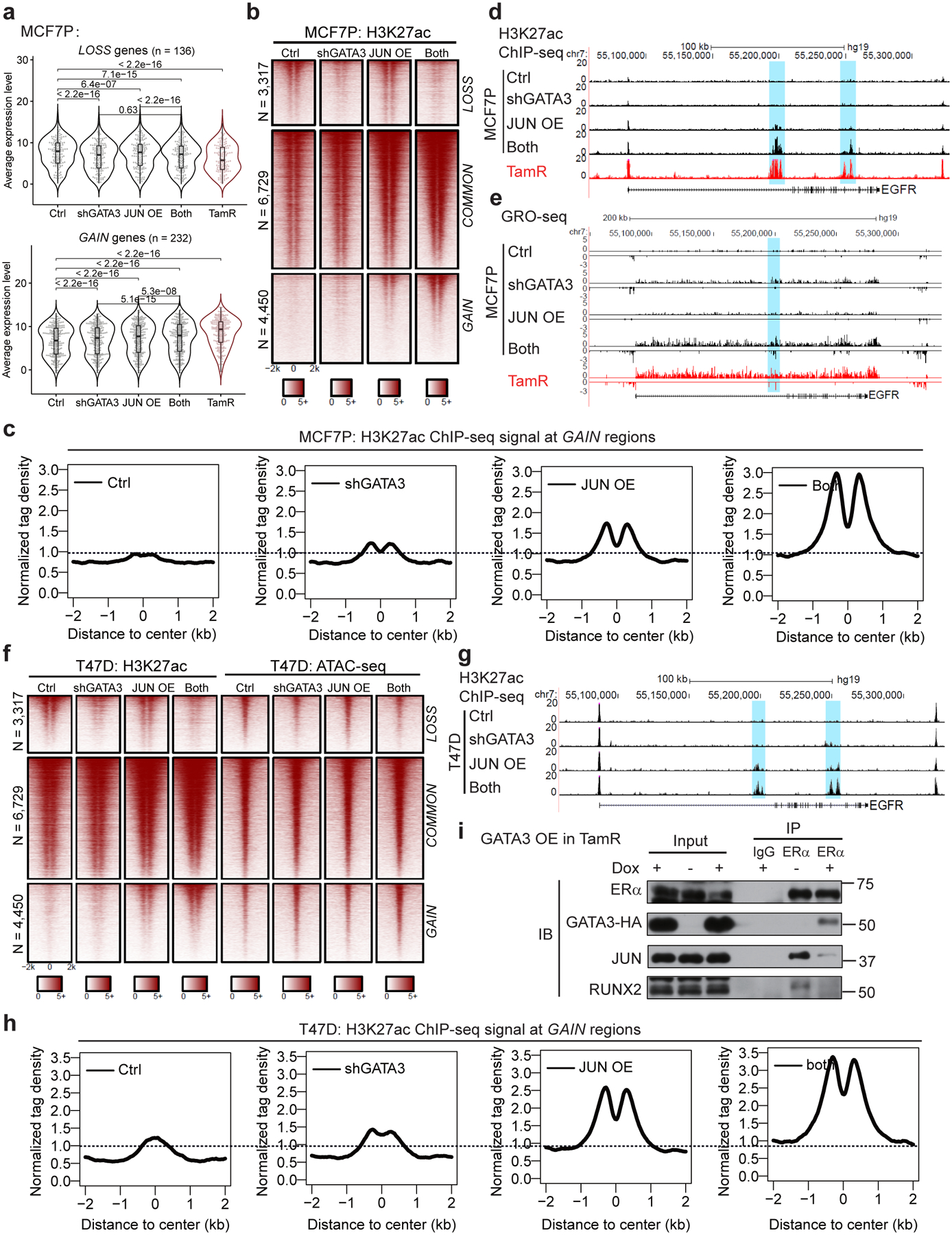
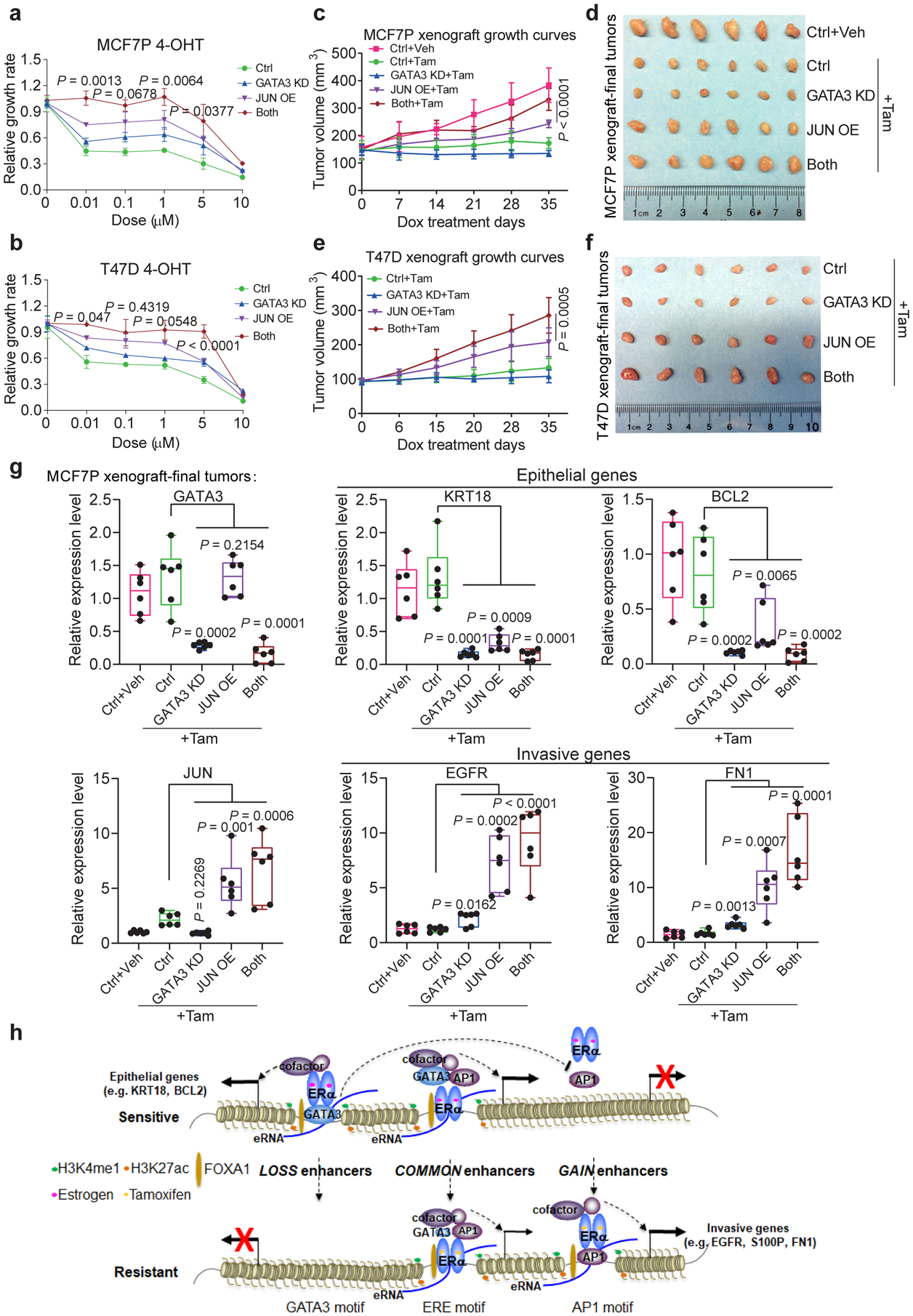
Acquired therapy resistance is a major problem for anticancer treatment, yet the underlying molecular mechanisms remain unclear. Using an established breast cancer cellular model, we show that endocrine resistance is associated with enhanced phenotypic plasticity, indicated by a general downregulation of luminal/epithelial differentiation markers and upregulation of basal/mesenchymal invasive markers. Consistently, similar gene expression changes are found in clinical breast tumours and patient-derived xenograft samples that are resistant to endocrine therapies. Mechanistically, the differential interactions between oestrogen receptor α and other oncogenic transcription factors, exemplified by GATA3 and AP1, drive global enhancer gain/loss reprogramming, profoundly altering breast cancer transcriptional programs. Our functional studies in multiple culture and xenograft models reveal a coordinated role of GATA3 and AP1 in re-organizing enhancer landscapes and regulating cancer phenotypes. Collectively, our study suggests that differential high-order assemblies of transcription factors on enhancers trigger genome-wide enhancer reprogramming, resulting in transcriptional transitions that promote tumour phenotypic plasticity and therapy resistance.
Figures
Comment in
-
A pliable ERα cistrome evades therapy.Nat Cell Biol. 2020 Jun;22(6):619-620. doi: 10.1038/s41556-020-0528-6. Nat Cell Biol. 2020. PMID: 32424274 No abstract available.
References
-
- Fu J & Jeffrey SS Transcriptomic signatures in breast cancer. Mol Biosyst 3, 466–472 (2007). - PubMed
-
- Murphy CG & Dickler MN Endocrine resistance in hormone-responsive breast cancer: mechanisms and therapeutic strategies. Endocr Relat Cancer 23, R337–352 (2016). - PubMed
-
- Musgrove EA & Sutherland RL Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9, 631–643 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
