

Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases
- PMID: 32424352
- PMCID: PMC7343625
- DOI: 10.1038/s41588-020-0628-z
Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases
Abstract
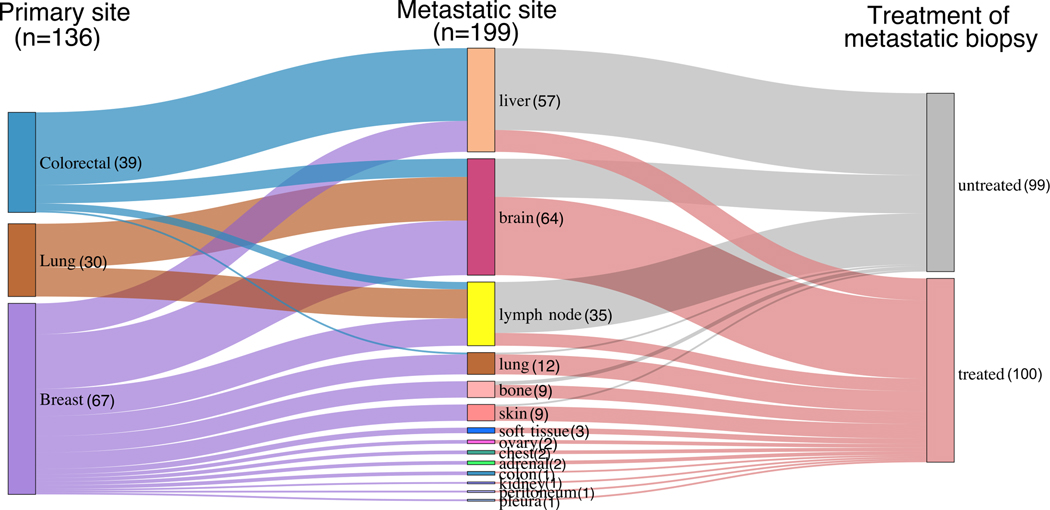
Metastasis is the primary cause of cancer-related deaths, but the natural history, clonal evolution and impact of treatment are poorly understood. We analyzed whole-exome sequencing (WES) data from 457 paired primary tumor and metastatic samples from 136 patients with breast, colorectal and lung cancer, including untreated (n = 99) and treated (n = 100) metastases. Treated metastases often harbored private 'driver' mutations, whereas untreated metastases did not, suggesting that treatment promotes clonal evolution. Polyclonal seeding was common in untreated lymph node metastases (n = 17 out of 29, 59%) and distant metastases (n = 20 out of 70, 29%), but less frequent in treated distant metastases (n = 9 out of 94, 10%). The low number of metastasis-private clonal mutations is consistent with early metastatic seeding, which we estimated occurred 2-4 years before diagnosis across these cancers. Furthermore, these data suggest that the natural course of metastasis is selectively relaxed relative to early tumorigenesis and that metastasis-private mutations are not drivers of cancer spread but instead associated with drug resistance.
Conflict of interest statement
Competing interests
C.C. is a scientific advisor to GRAIL and reports stock options as well as consulting for GRAIL and Genentech. Z.H., Z.L., Z.M. have no conflicts of interest to report.
Figures














References
-
- Talmadge JE, Wolman SR & Fidler IJ Evidence for the clonal origin of spontaneous metastases. Science 217, 361–3 (1982). - PubMed
-
- Yamamoto N. et al. Determination of clonality of metastasis by cell-specific color-coded fluorescent-protein imaging. Cancer Res 63, 7785–90 (2003). - PubMed
-
- Huang Y. et al. Multilayered molecular profiling supported the monoclonal origin of metastatic renal cell carcinoma. Int J Cancer 135, 78–87 (2014). - PubMed
Methods-only references
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
