

De novo mutations in FBRSL1 cause a novel recognizable malformation and intellectual disability syndrome
- PMID: 32424618
- PMCID: PMC7519918
- DOI: 10.1007/s00439-020-02175-x
De novo mutations in FBRSL1 cause a novel recognizable malformation and intellectual disability syndrome
Abstract
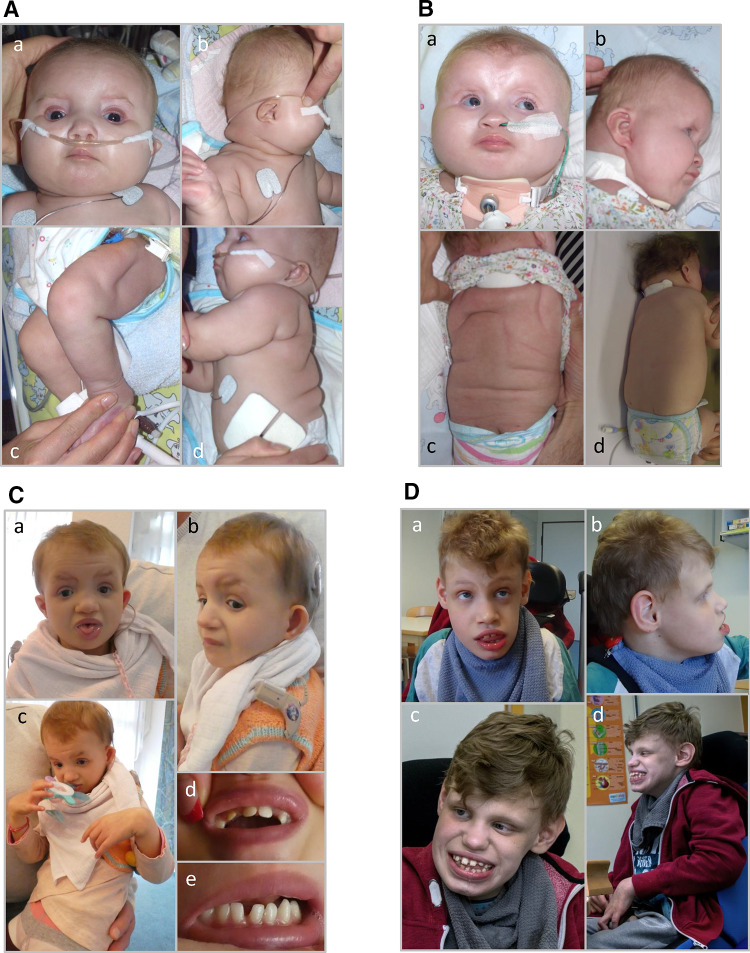
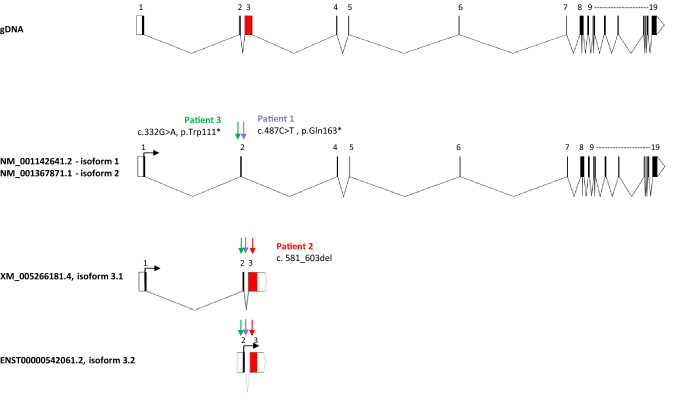
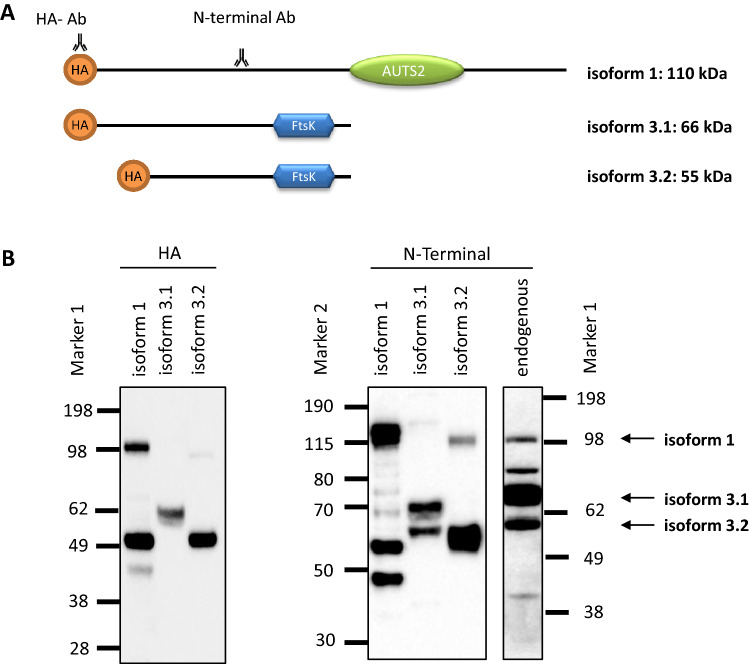
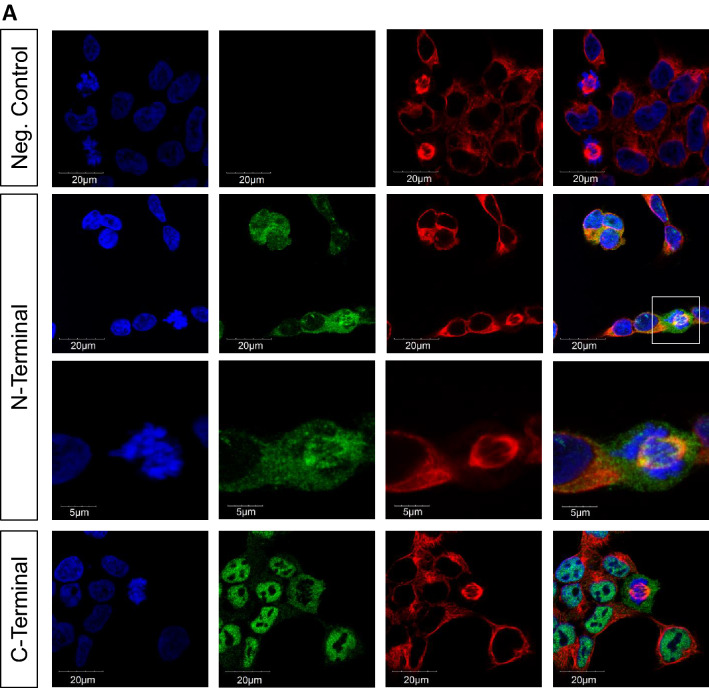
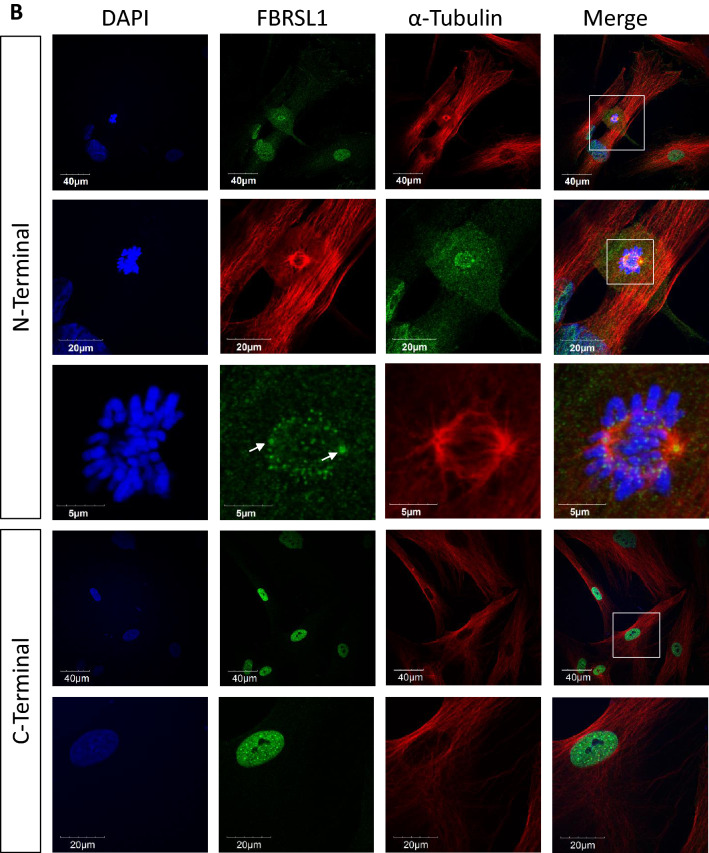
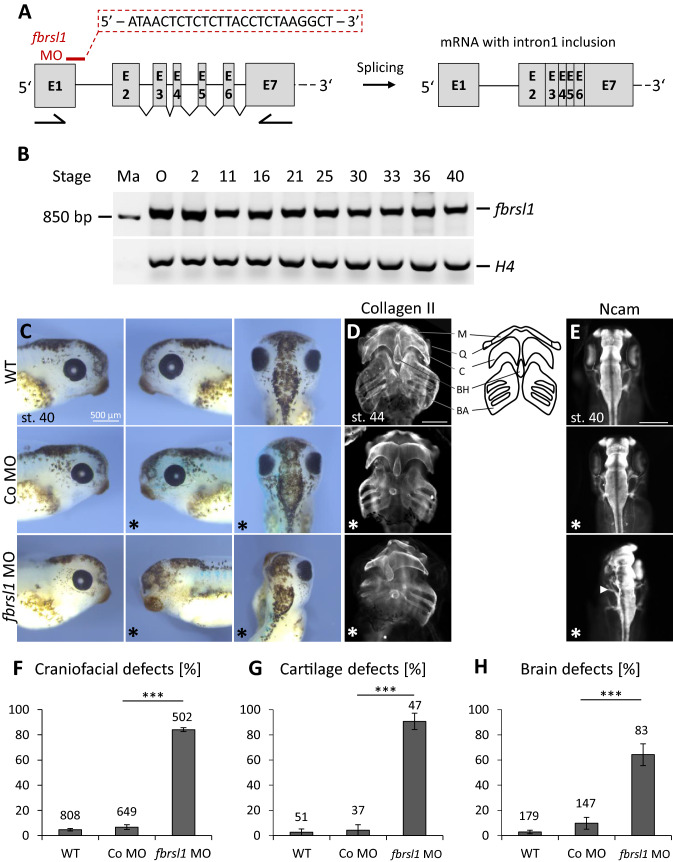
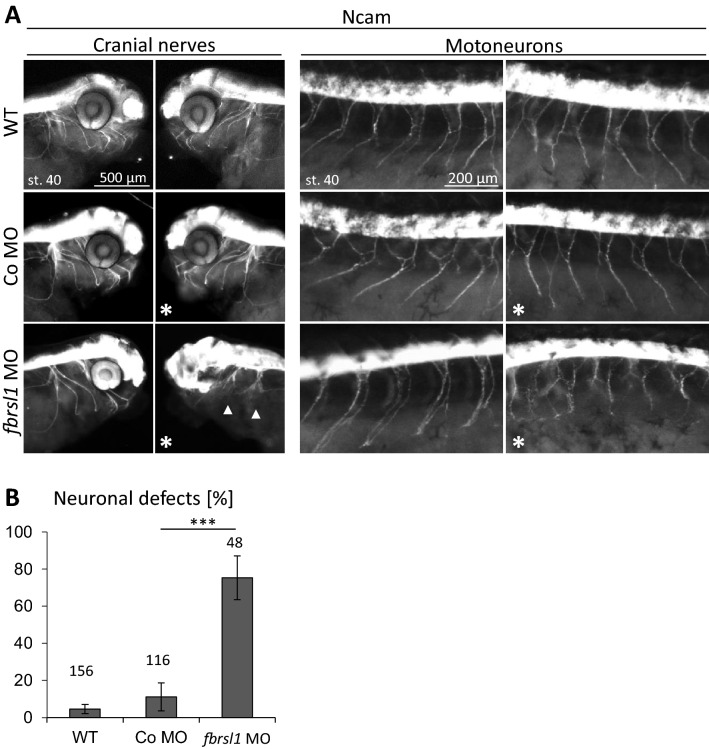
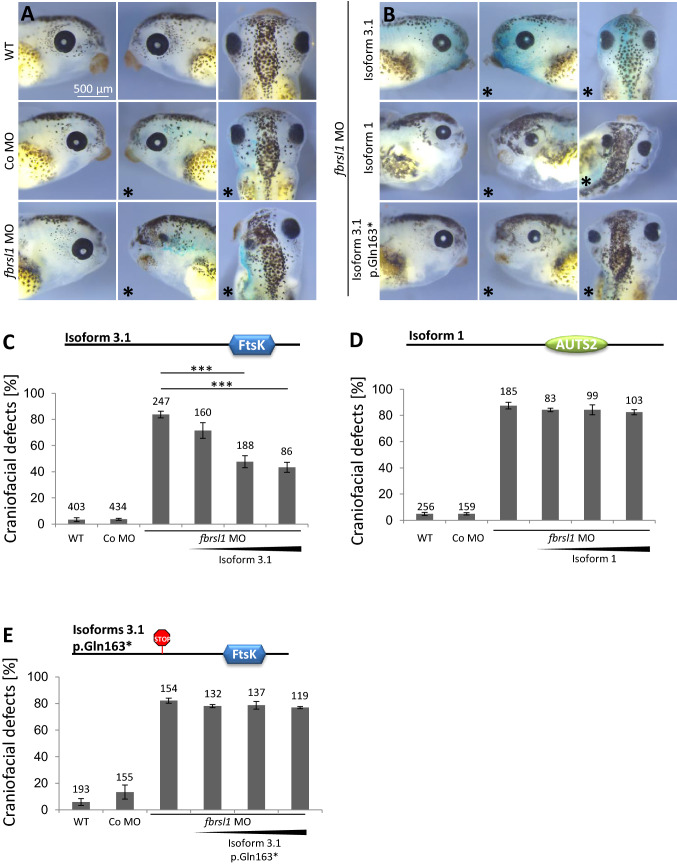
We report truncating de novo variants in specific exons of FBRSL1 in three unrelated children with an overlapping syndromic phenotype with respiratory insufficiency, postnatal growth restriction, microcephaly, global developmental delay and other malformations. The function of FBRSL1 is largely unknown. Interestingly, mutations in the FBRSL1 paralogue AUTS2 lead to an intellectual disability syndrome (AUTS2 syndrome). We determined human FBRSL1 transcripts and describe protein-coding forms by Western blot analysis as well as the cellular localization by immunocytochemistry stainings. All detected mutations affect the two short N-terminal isoforms, which show a ubiquitous expression in fetal tissues. Next, we performed a Fbrsl1 knockdown in Xenopus laevis embryos to explore the role of Fbrsl1 during development and detected craniofacial abnormalities and a disturbance in neurite outgrowth. The aberrant phenotype in Xenopus laevis embryos could be rescued with a human N-terminal isoform, while the long isoform and the N-terminal isoform containing the mutation p.Gln163* isolated from a patient could not rescue the craniofacial defects caused by Fbrsl1 depletion. Based on these data, we propose that the disruption of the validated N-terminal isoforms of FBRSL1 at critical timepoints during embryogenesis leads to a hitherto undescribed complex neurodevelopmental syndrome.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Beunders G, Voorhoeve E, Golzio C, Pardo LM, Rosenfeld JA, Talkowski ME, Simonic I, Lionel AC, Vergult S, Pyatt RE, van de Kamp J, Nieuwint A, Weiss MM, Rizzu P, Verwer LENI, van Spaendonk RML, Shen Y, Wu B-l, Yu T, Yu Y, Chiang C, Gusella JF, Lindgren AM, Morton CC, van Binsbergen E, Bulk S, van Rossem E, Vanakker O, Armstrong R, Park S-M, Greenhalgh L, Maye U, Neill NJ, Abbott KM, Sell S, Ladda R, Farber DM, Bader PI, Cushing T, Drautz JM, Konczal L, Nash P, de Los Reyes E, Carter MT, Hopkins E, Marshall CR, Osborne LR, Gripp KW, Thrush DL, Hashimoto S, Gastier-Foster JM, Astbury C, Ylstra B, Meijers-Heijboer H, Posthuma D, Menten B, Mortier G, Scherer SW, Eichler EE, Girirajan S, Katsanis N, Groffen AJ, Sistermans EA. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet. 2013;92(2):210–220. doi: 10.1016/j.ajhg.2012.12.011. - DOI - PMC - PubMed
-
- Beunders G, de Munnik SA, Van der Aa N, Ceulemans B, Voorhoeve E, Groffen AJ, Nillesen WM, Meijers-Heijboer EJ, Frank Kooy R, Yntema HG, Sistermans EA. Two male adults with pathogenic AUTS2 variants, including a two-base pair deletion, further delineate the AUTS2 syndrome. Eur J Hum Genet. 2015;23(6):803–807. doi: 10.1038/ejhg.2014.173. - DOI - PMC - PubMed
-
- Beunders G, van de Kamp J, Vasudevan P, Morton J, Smets K, Kleefstra T, de Munnik SA, Schuurs-Hoeijmakers J, Ceulemans B, Zollino M, Hoffjan S, Wieczorek S, So J, Mercer L, Walker T, Velsher L, Parker MJ, Magee AC, Elffers B, Kooy RF, Yntema HG, Meijers-Heijboer EJ, Sistermans EA. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J Med Genet. 2016;53(8):523–532. doi: 10.1136/jmedgenet-2015-103601. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
