

Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease
- PMID: 32425433
- PMCID: PMC7220963
- DOI: 10.6515/ACS.202005_36(3).20200213A
Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease
Abstract
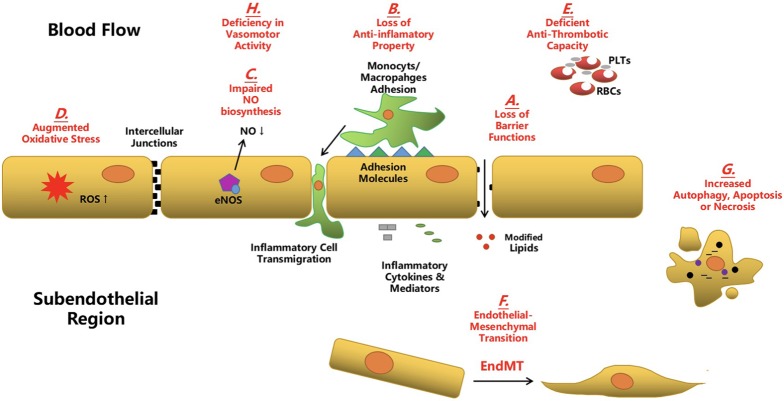
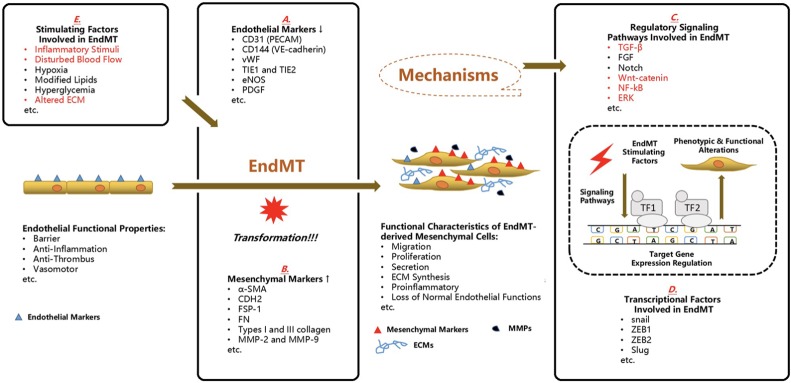
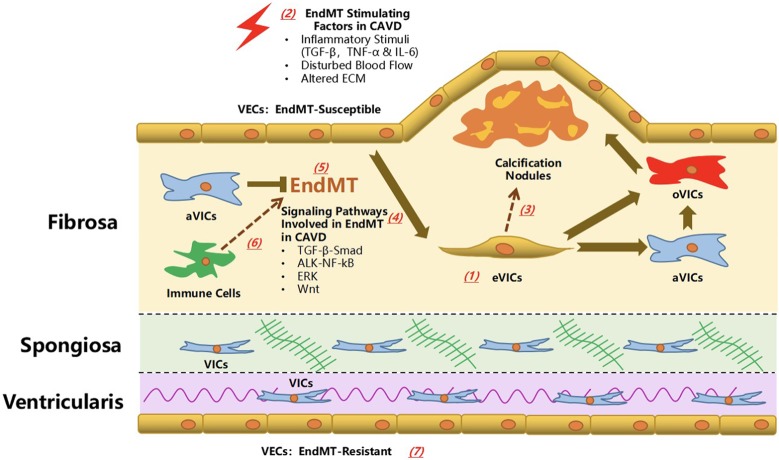
Calcific aortic valve disease (CAVD) represents a significant threat to cardiovascular health worldwide, and the incidence of this sclerocalcific valve disease has rapidly increased along with a rise in life expectancy. Compelling evidence has suggested that CAVD is an actively and finely regulated pathophysiological process even though it has been referred to as "degenerative" for decades. A striking similarity has been noted in the etiopathogenesis between CAVD and atherosclerosis, a classical proliferative sclerotic vascular disease.1 Nevertheless, pharmaceutical trials that attempted to target inflammation and dyslipidemia have produced disappointing results in CAVD. While senescence is a well-documented risk factor, the sophisticated regulatory networks have not been adequately explored underlying the aberrant calcification and osteogenesis in CAVD. Valvular endothelial cells (VECs), a type of resident effector cells in aortic leaflets, are crucial in maintaining valvular integrity and homeostasis, and dysfunctional VECs are a major contributor to disease initiation and progression. Accumulating evidence suggests that VECs undergo a phenotypic and functional transition to mesenchymal or fibroblast-like cells in CAVD, a process known as the endothelial-to-mesenchymal transition (EndMT) process. The relevance of this transition in CAVD has recently drawn great interest due to its importance in both valve genesis at an embryonic stage and CAVD development at an adult stage. Hence EndMT might be a valuable diagnostic and therapeutic target for disease prevention and treatment. This mini-review summarized the relevant literature that delineates the EndMT process and the underlying regulatory networks involved in CAVD.
Keywords: Calcific aortic valve disease; Endothelial-to-mesenchymal transition; Regulatory network; Valvular endothelial cell.
Figures
References
-
- Nkomo VT, Gardin JM, Skelton TN, et al. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. - PubMed
-
- Shen M, Tastet L, Bergler-Klein J, et al. Blood, tissue and imaging biomarkers in calcific aortic valve stenosis: past, present and future. Curr Opin Cardiol. 2018;33:125–133. - PubMed
Publication types
LinkOut - more resources
Full Text Sources