

Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the CDH23 Gene Causing Usher Syndrome Type ID in a Chinese Patient
- PMID: 32425987
- PMCID: PMC7204213
- DOI: 10.3389/fgene.2020.00422
Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the CDH23 Gene Causing Usher Syndrome Type ID in a Chinese Patient
Abstract
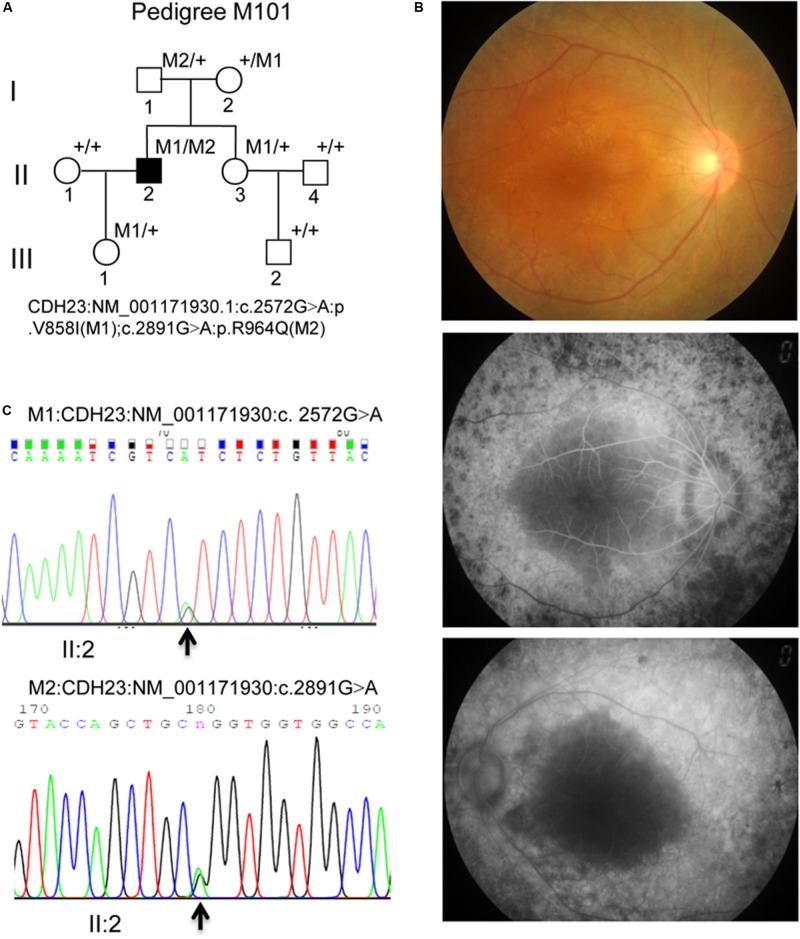
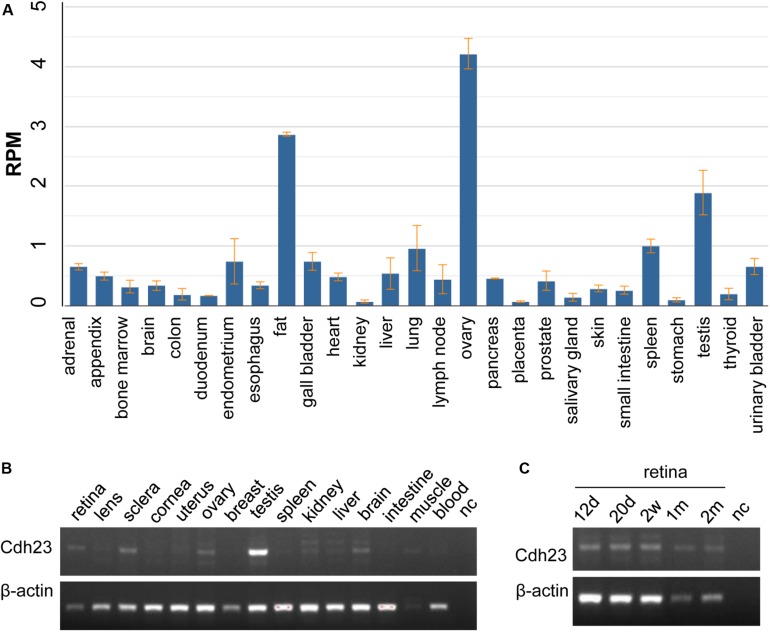
Usher syndrome includes a group of genetically and clinically heterogeneous autosomal recessive diseases, such as retinitis pigmentosa (RP) and sensorineural hearing loss. Usher syndrome type I (USHI) is characterized by profound hearing impairment beginning at birth, vestibular dysfunction, and unintelligible speech in addition to RP. The relationships between the Usher syndrome causing genes and the resultant phenotypes of Usher syndrome have not yet been fully elucidated. In the present study, we recruited a Chinese family with Usher syndrome and conducted paneled next-generation sequencing, Sanger sequencing, segregation analysis, and expression profile analysis. The functional effects of the identified cadherin-related 23 (CDH23) pathogenic variants were analyzed. The M101 pedigree consisted of a proband and seven family members, and the proband was a 39-year-old Chinese male who claimed that he first began to experience night blindness 11 years ago. We revealed novel, missense compound heterozygous variants c. 2572G > A (p.V858I) and c. 2891G > A (p.R964Q) in the CDH23 gene, which co-segregated with the disease phenotype causing Usher syndrome type ID (USH1D) in this Chinese pedigree. CDH23 mRNA was highly expressed in the retina, and this protein was highly conserved as revealed by the comparison of Homo sapiens CDH23 with those from nine other species. This is the first study to identify the novel, missense compound heterozygous variants c. 2572G > A (p.V858I) and c.2891G > A (p.R964Q) of CDH23, which might cause USH1D in the studied Chinese family, thereby extending CDH23 mutation spectra. Identifying CDH23 pathogenic variants should help in the detailed phenotypic characterization of USH1D.
Keywords: CDH23 gene; Usher syndrome type ID; genotype/phenotype correlation; missense mutation; targeted next-generation sequencing.
Copyright © 2020 Zhang, Cheng, Zhou, Khan, Fu, Duan, Sun, Lv and Fu.
Figures

References
-
- An J., Yang J., Wang Y., Wang Y., Xu B., Xie G., et al. (2019). Targeted next generation sequencing revealed a novel homozygous loss-of-function mutation in ILDR1 gene causes autosomal recessive nonsyndromic sensorineural hearing loss in a Chinese family. Front. Genet. 10:1. 10.3389/fgene.2019.00001 - DOI - PMC - PubMed
-
- Bork J. M., Peters L. M., Riazuddin S., Bernstein S. L., Ahmed Z. M., Ness S. L., et al. (2001). Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68 26–37. 10.1086/316954 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases