

Molecular networking in the neuronal ceroid lipofuscinoses: insights from mammalian models and the social amoeba Dictyostelium discoideum
- PMID: 32430003
- PMCID: PMC7238602
- DOI: 10.1186/s12929-020-00653-y
Molecular networking in the neuronal ceroid lipofuscinoses: insights from mammalian models and the social amoeba Dictyostelium discoideum
Erratum in
-
Correction to: Molecular networking in the neuronal ceroid lipofuscinoses: insights from mammalian models and the social amoeba Dictyostelium discoideum.J Biomed Sci. 2020 Sep 21;27(1):94. doi: 10.1186/s12929-020-00686-3. J Biomed Sci. 2020. PMID: 32951590 Free PMC article.
Abstract
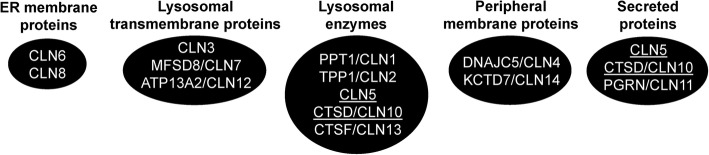
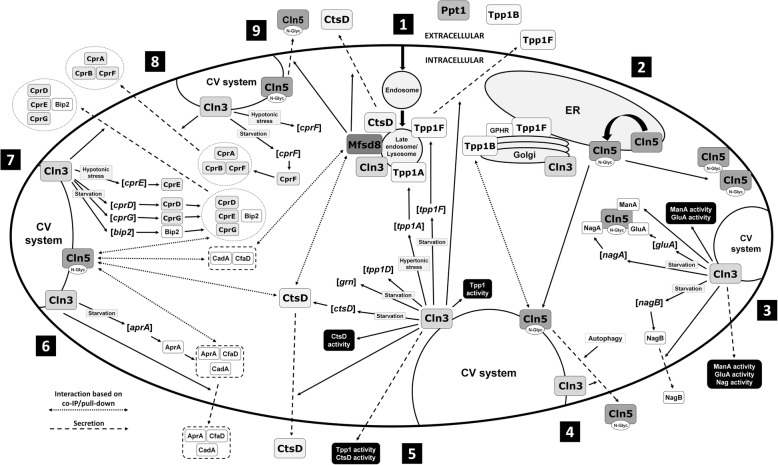
The neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease, belong to a family of neurological disorders that cause blindness, seizures, loss of motor function and cognitive ability, and premature death. There are 13 different subtypes of NCL that are associated with mutations in 13 genetically distinct genes (CLN1-CLN8, CLN10-CLN14). Similar clinical and pathological profiles of the different NCL subtypes suggest that common disease mechanisms may be involved. As a result, there have been many efforts to determine how NCL proteins are connected at the cellular level. A main driving force for NCL research has been the utilization of mammalian and non-mammalian cellular models to study the mechanisms underlying the disease. One non-mammalian model that has provided significant insight into NCL protein function is the social amoeba Dictyostelium discoideum. Accumulated data from Dictyostelium and mammalian cells show that NCL proteins display similar localizations, have common binding partners, and regulate the expression and activities of one another. In addition, genetic models of NCL display similar phenotypes. This review integrates findings from Dictyostelium and mammalian models of NCL to highlight our understanding of the molecular networking of NCL proteins. The goal here is to help set the stage for future work to reveal the cellular mechanisms underlying the NCLs.
Keywords: Batten disease; Dictyostelium discoideum; Neurodegeneration; Neuronal ceroid lipofuscinosis; Molecular networking.
Conflict of interest statement
None.
Figures
References
-
- Cárcel-Trullols J, Kovács AD, Pearce DA. Cell biology of the NCL proteins: what they do and don’t do. Biochim Biophys Acta. 1852;2015:2242–2255. - PubMed
-
- Butz ES, Chandrachud U, Mole SE, Cotman SL. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol basis Dis. 2019;1:165571. - PubMed
-
- Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. 1852;2015:2262–2266. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
