

Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: effect on imatinib resistance and stem cells
- PMID: 32430012
- PMCID: PMC7236970
- DOI: 10.1186/s13148-020-00839-z
Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: effect on imatinib resistance and stem cells
Abstract
Background: Chronic myeloid leukemia (CML) pathogenesis is mainly driven by the oncogenic breakpoint cluster region-Abelson murine leukemia viral oncogene homolog 1 (BCR-ABL) fusion protein. Since BCR-ABL displays abnormal constitutive tyrosine kinase activity, therapies using tyrosine kinase inhibitors (TKis) such as imatinib represent a major breakthrough for the outcome of CML patients. Nevertheless, the development of TKi resistance and the persistence of leukemia stem cells (LSCs) remain barriers to cure the disease, justifying the development of novel therapeutic approaches. Since the activity of histone deacetylase (HDAC) is deregulated in numerous cancers including CML, pan-HDAC inhibitors may represent promising therapeutic regimens for the treatment of CML cells in combination with TKi.
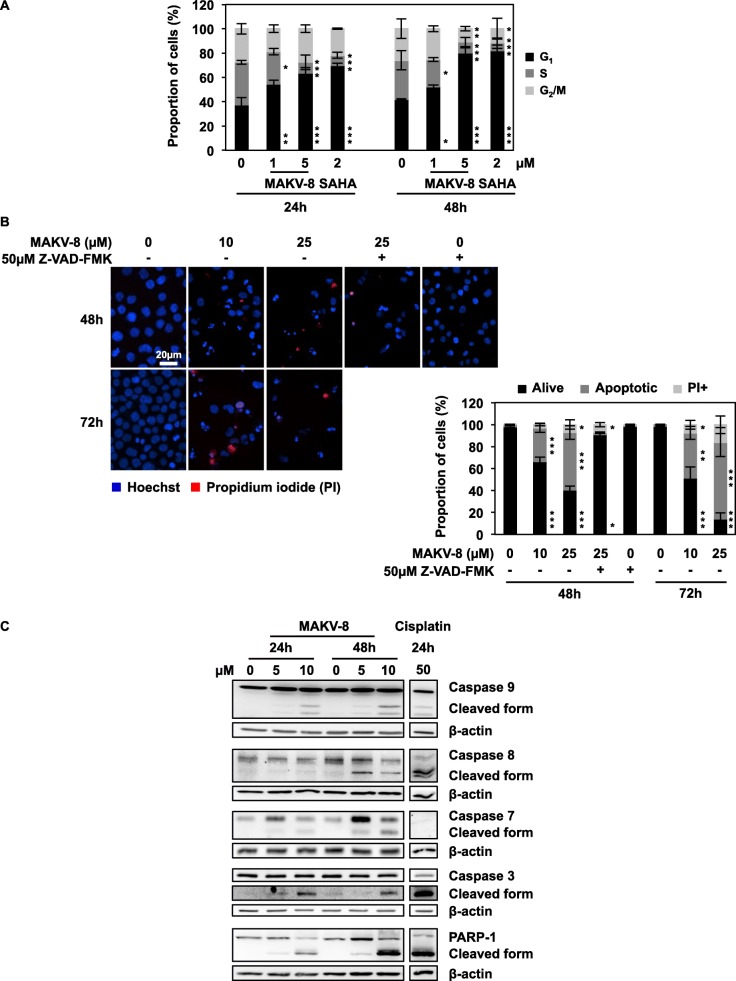
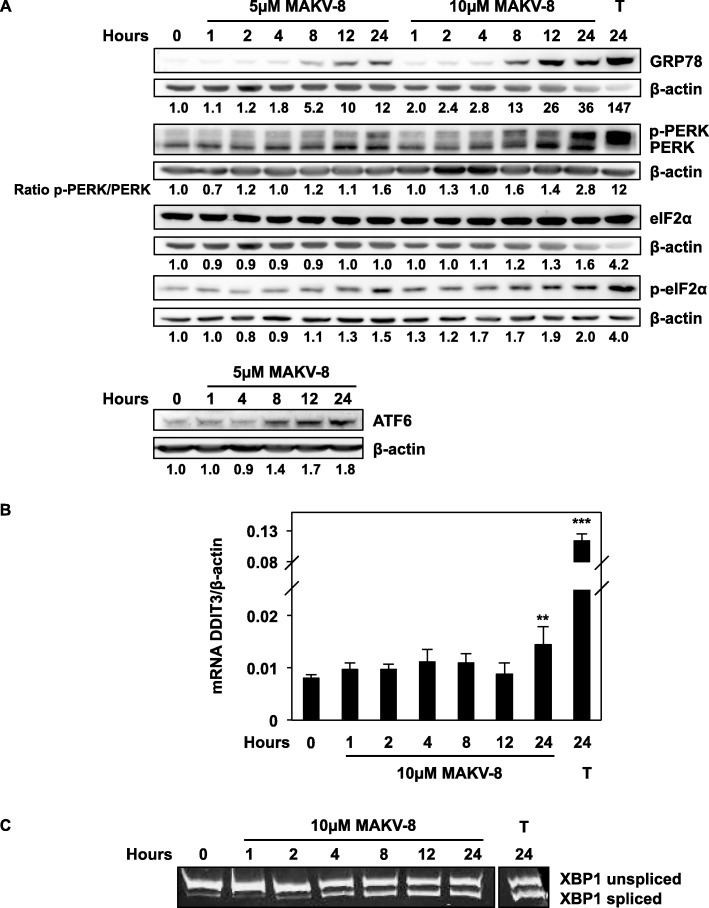
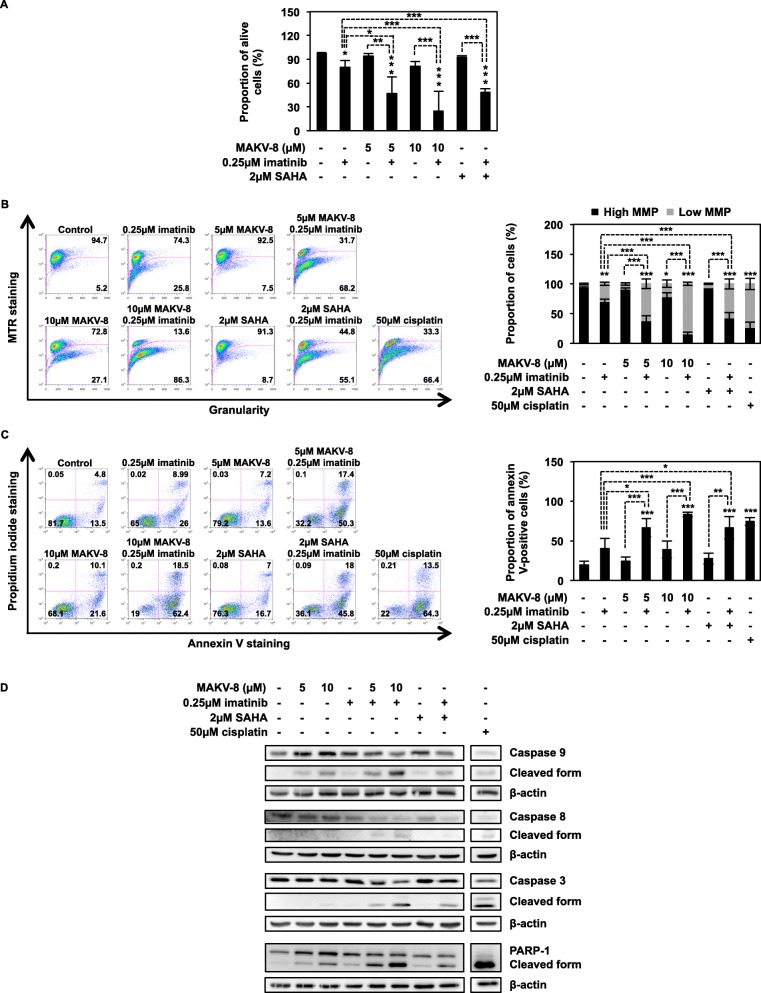
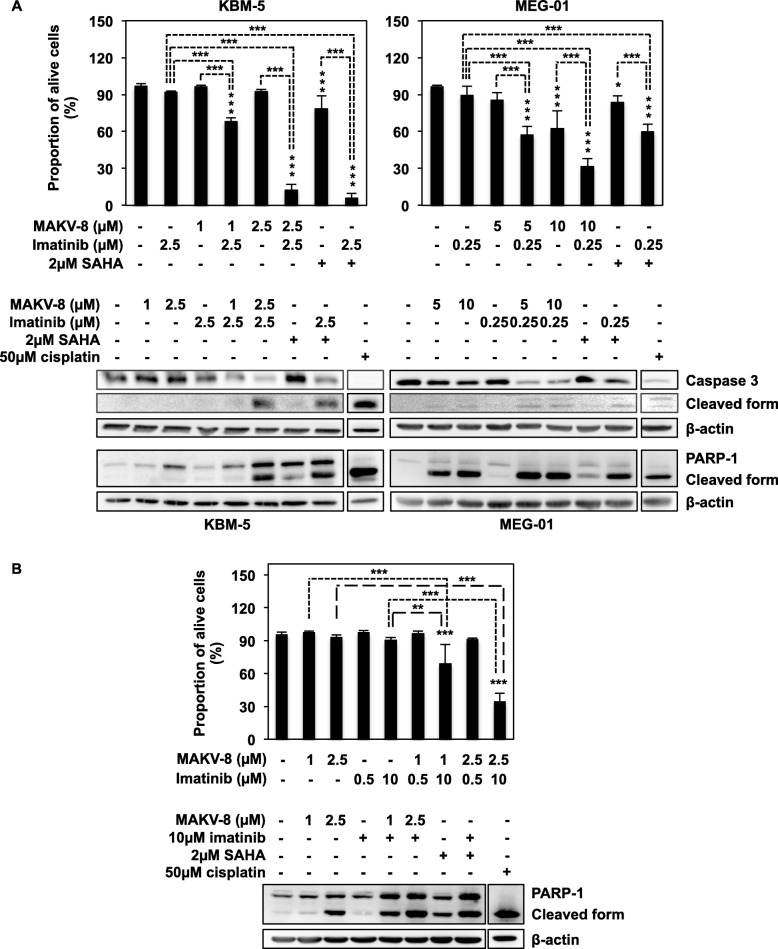
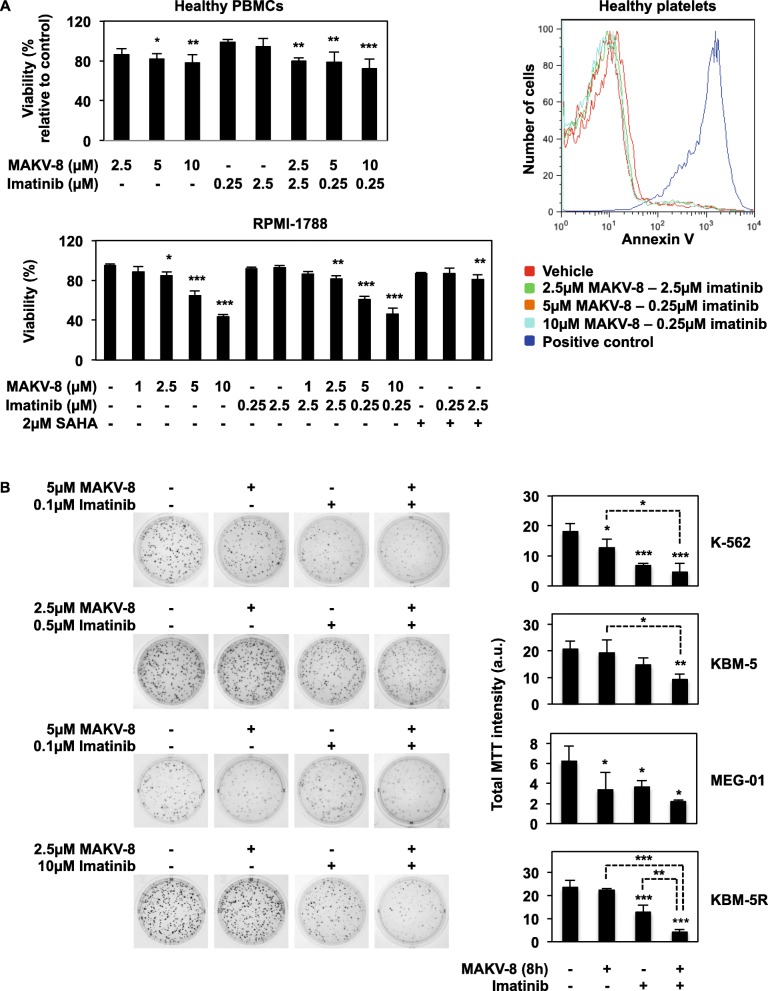
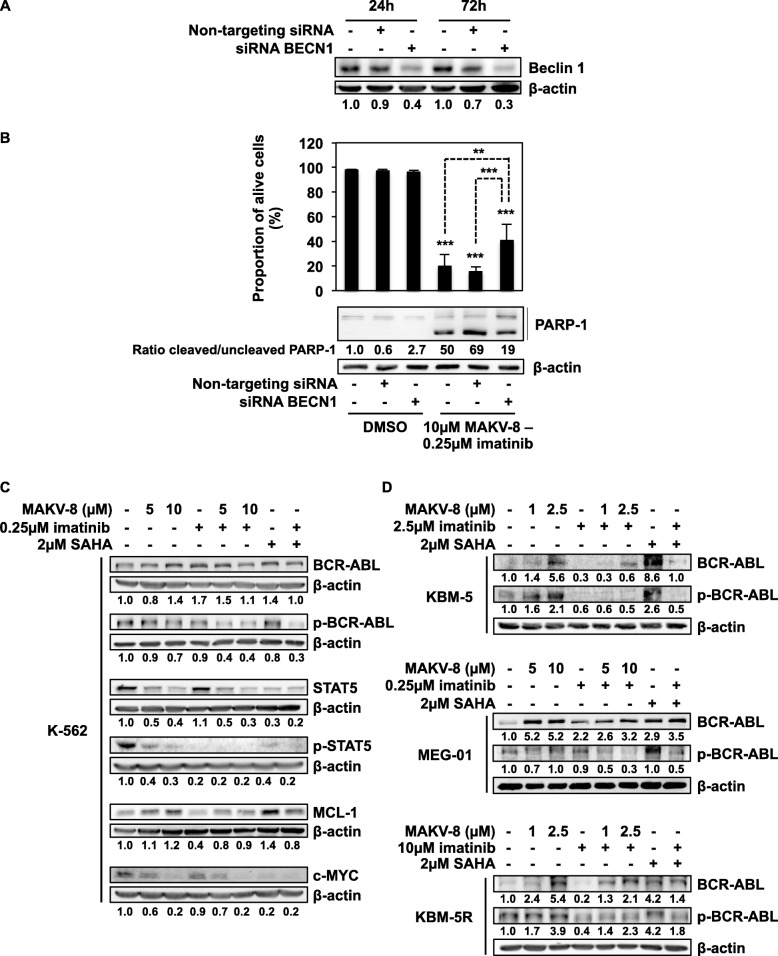
Results: We assessed the anti-leukemic activity of a novel hydroxamate-based pan-HDAC inhibitor MAKV-8, which complied with the Lipinski's "rule of five," in various CML cells alone or in combination with imatinib. We validated the in vitro HDAC-inhibitory potential of MAKV-8 and demonstrated efficient binding to the ligand-binding pocket of HDAC isoenzymes. In cellulo, MAKV-8 significantly induced target protein acetylation, displayed cytostatic and cytotoxic properties, and triggered concomitant ER stress/protective autophagy leading to canonical caspase-dependent apoptosis. Considering the specific upregulation of selected HDACs in LSCs from CML patients, we investigated the differential toxicity of a co-treatment with MAKV-8 and imatinib in CML versus healthy cells. We also showed that beclin-1 knockdown prevented MAKV-8-imatinib combination-induced apoptosis. Moreover, MAKV-8 and imatinib co-treatment synergistically reduced BCR-ABL-related signaling pathways involved in CML cell growth and survival. Since our results showed that LSCs from CML patients overexpressed c-MYC, importantly MAKV-8-imatinib co-treatment reduced c-MYC levels and the LSC population. In vivo, tumor growth of xenografted K-562 cells in zebrafish was completely abrogated upon combined treatment with MAKV-8 and imatinib.
Conclusions: Collectively, the present findings show that combinations HDAC inhibitor-imatinib are likely to overcome drug resistance in CML pathology.
Keywords: Apoptosis; Autophagy; Computational docking; Endoplasmic reticulum stress; Epigenetic regulation; Tyrosine kinase inhibitor.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
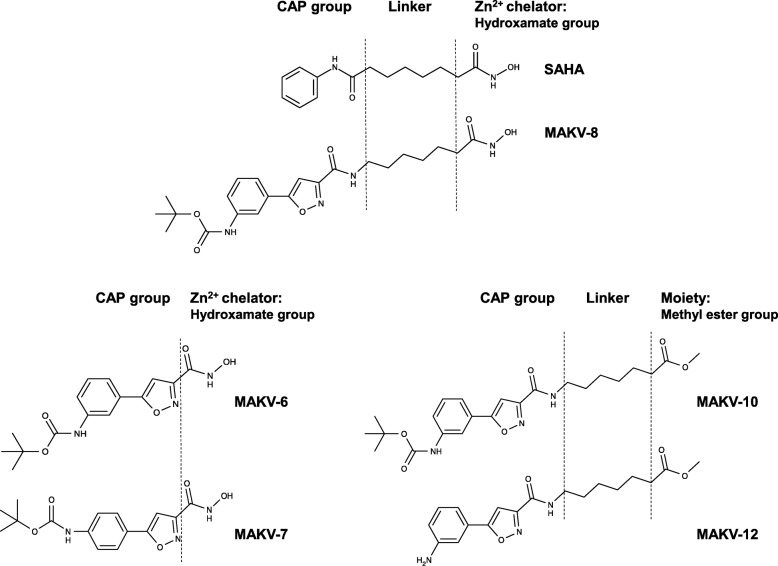
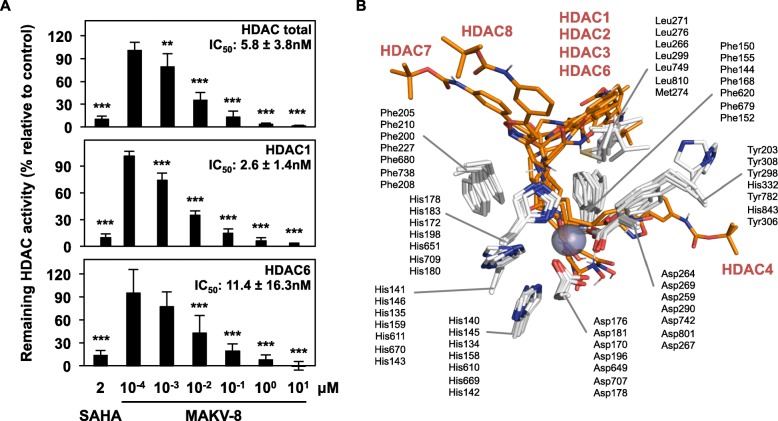
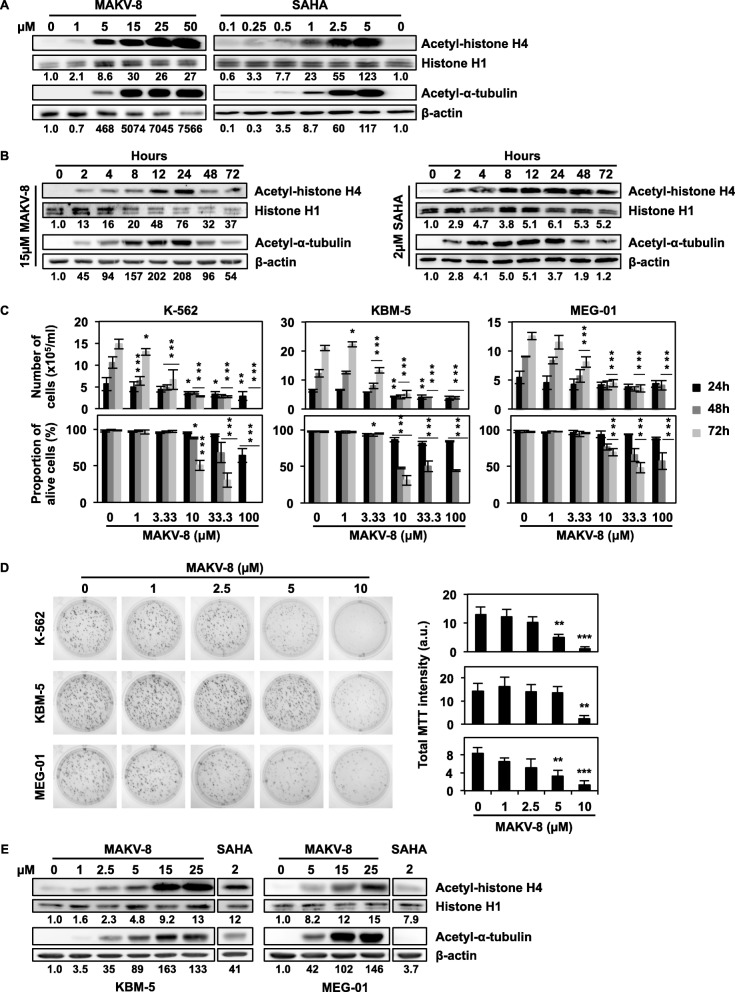
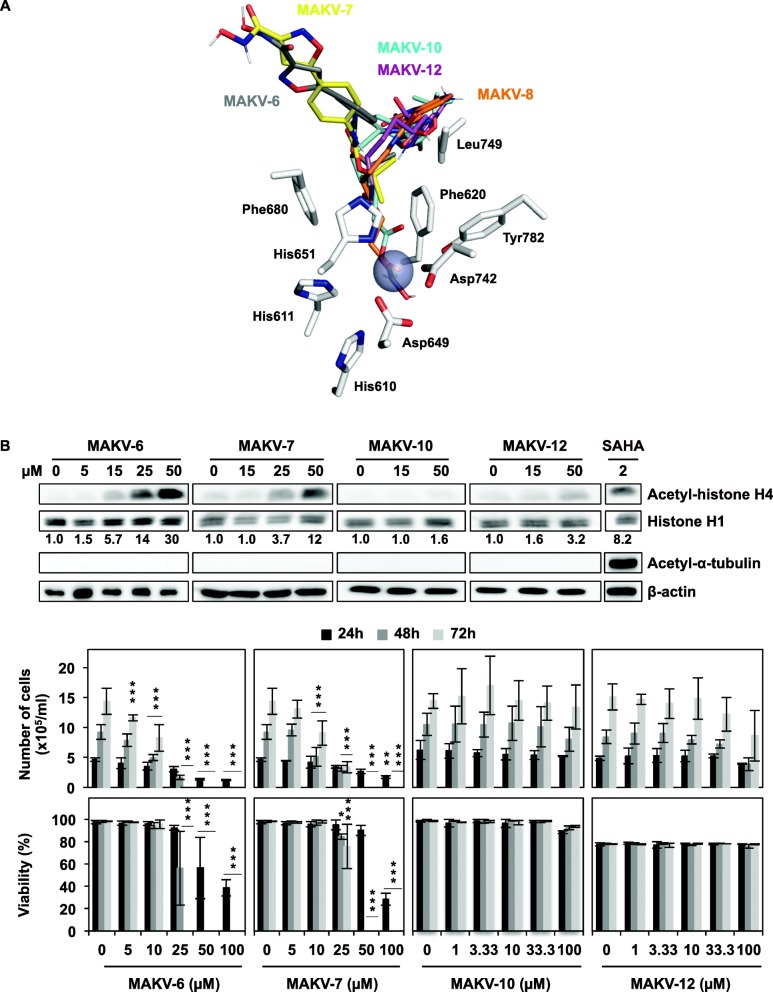



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
