

EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer
- PMID: 32430388
- PMCID: PMC7416460
- DOI: 10.1158/2159-8290.CD-20-0187
EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer
Abstract
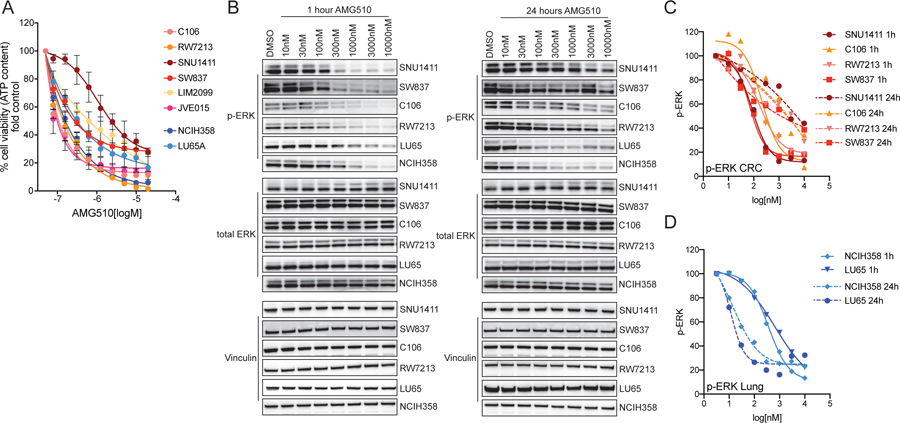
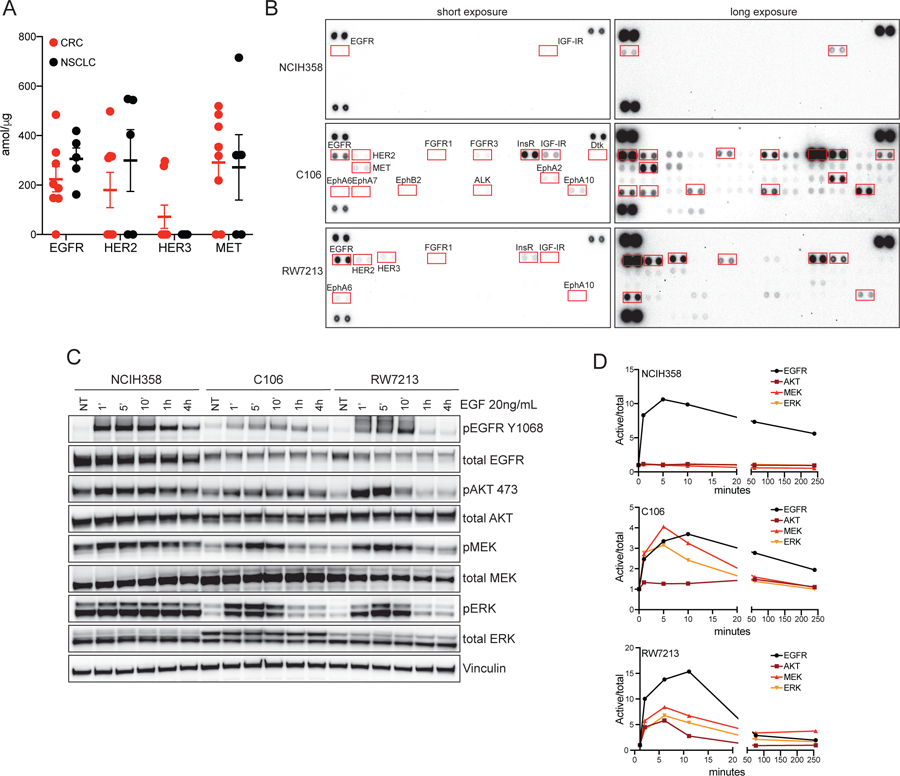
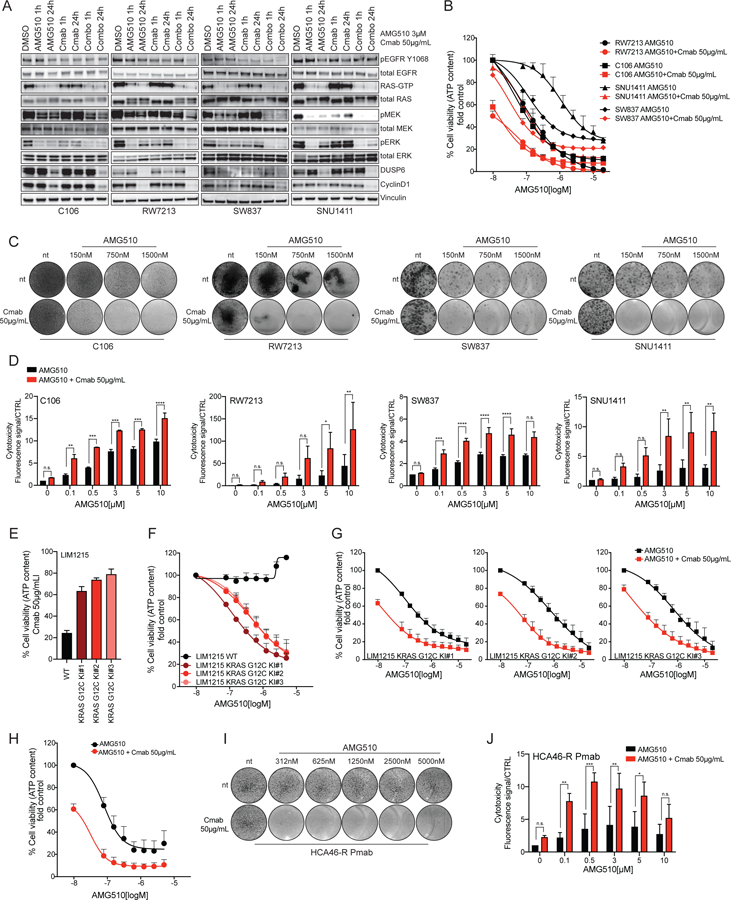
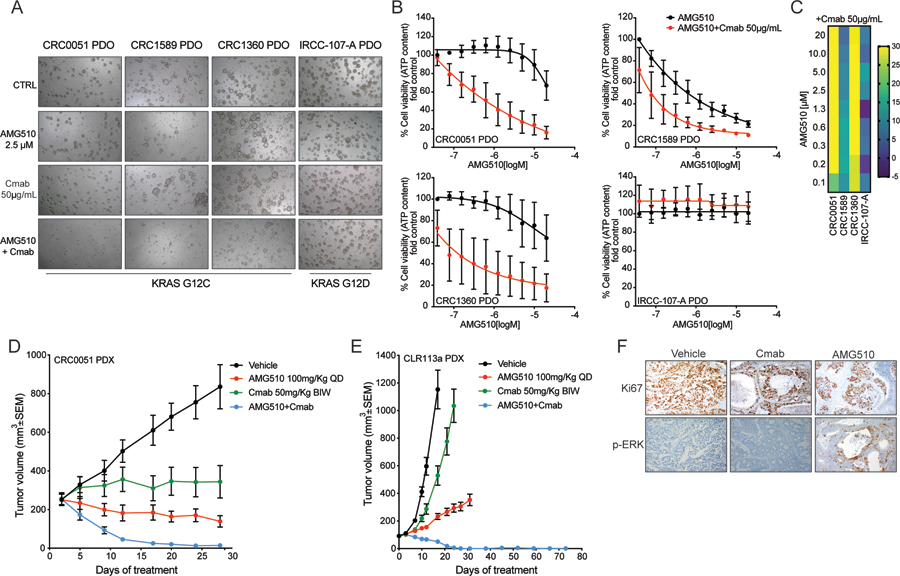
Most patients with KRAS G12C-mutant non-small cell lung cancer (NSCLC) experience clinical benefit from selective KRASG12C inhibition, whereas patients with colorectal cancer bearing the same mutation rarely respond. To investigate the cause of the limited efficacy of KRASG12C inhibitors in colorectal cancer, we examined the effects of AMG510 in KRAS G12C colorectal cancer cell lines. Unlike NSCLC cell lines, KRAS G12C colorectal cancer models have high basal receptor tyrosine kinase (RTK) activation and are responsive to growth factor stimulation. In colorectal cancer lines, KRASG12C inhibition induces higher phospho-ERK rebound than in NSCLC cells. Although upstream activation of several RTKs interferes with KRASG12C blockade, we identify EGFR signaling as the dominant mechanism of colorectal cancer resistance to KRASG12C inhibitors. The combinatorial targeting of EGFR and KRASG12C is highly effective in colorectal cancer cells and patient-derived organoids and xenografts, suggesting a novel therapeutic strategy to treat patients with KRAS G12C colorectal cancer. SIGNIFICANCE: The efficacy of KRASG12C inhibitors in NSCLC and colorectal cancer is lineage-specific. RTK dependency and signaling rebound kinetics are responsible for sensitivity or resistance to KRASG12C inhibition in colorectal cancer. EGFR and KRASG12C should be concomitantly inhibited to overcome resistance to KRASG12C blockade in colorectal tumors.See related commentary by Koleilat and Kwong, p. 1094.This article is highlighted in the In This Issue feature, p. 1079.
©2020 American Association for Cancer Research.
Conflict of interest statement
conflicts of interest
All the other authors declare no conflicts of interests.
Figures
Comment in
-
AMG 510 Shows Activity beyond NSCLC.Cancer Discov. 2020 Aug;10(8):1084-1085. doi: 10.1158/2159-8290.CD-NB2020-061. Epub 2020 Jun 15. Cancer Discov. 2020. PMID: 32540954
-
Same Name, Different Game: EGFR Drives Intrinsic KRASG12C Inhibitor Resistance in Colorectal Cancer.Cancer Discov. 2020 Aug;10(8):1094-1096. doi: 10.1158/2159-8290.CD-20-0612. Cancer Discov. 2020. PMID: 32747369
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
