

Genetics of progression from MDS to secondary leukemia
- PMID: 32430504
- PMCID: PMC7332895
- DOI: 10.1182/blood.2019000942
Genetics of progression from MDS to secondary leukemia
Abstract
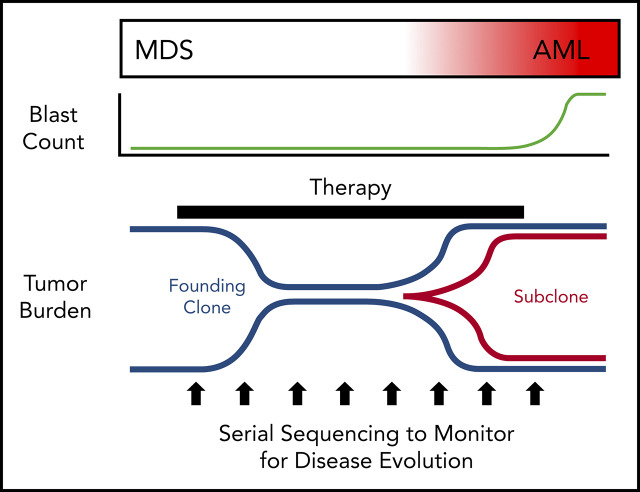
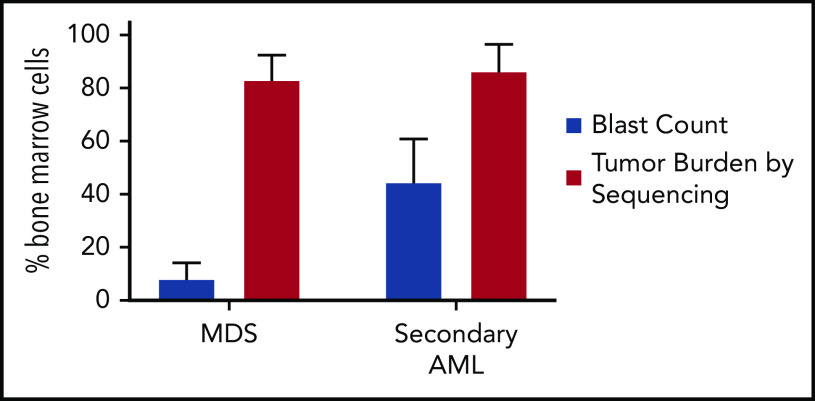
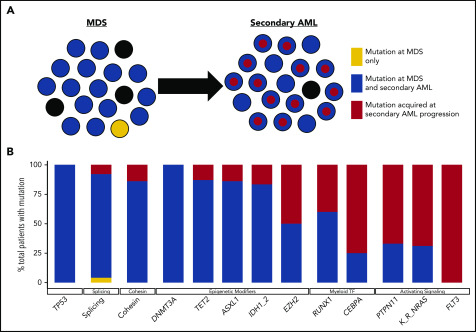
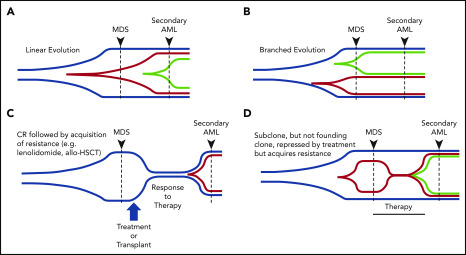
Our understanding of the genetics of acute myeloid leukemia (AML) development from myelodysplastic syndrome (MDS) has advanced significantly as a result of next-generation sequencing technology. Although differences in cell biology and maturation exist between MDS and AML secondary to MDS, these 2 diseases are genetically related. MDS and secondary AML cells harbor mutations in many of the same genes and functional categories, including chromatin modification, DNA methylation, RNA splicing, cohesin complex, transcription factors, cell signaling, and DNA damage, confirming that they are a disease continuum. Differences in the frequency of mutated genes in MDS and secondary AML indicate that the order of mutation acquisition is not random during progression. In almost every case, disease progression is associated with clonal evolution, typically defined by the expansion or emergence of a subclone with a unique set of mutations. Monitoring tumor burden and clonal evolution using sequencing provides advantages over using the blast count, which underestimates tumor burden, and could allow for early detection of disease progression prior to clinical deterioration. In this review, we outline advances in the study of MDS to secondary AML progression, with a focus on the genetics of progression, and discuss the advantages of incorporating molecular genetic data in the diagnosis, classification, and monitoring of MDS to secondary AML progression. Because sequencing is becoming routine in the clinic, ongoing research is needed to define the optimal assay to use in different clinical situations and how the data can be used to improve outcomes for patients with MDS and secondary AML.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
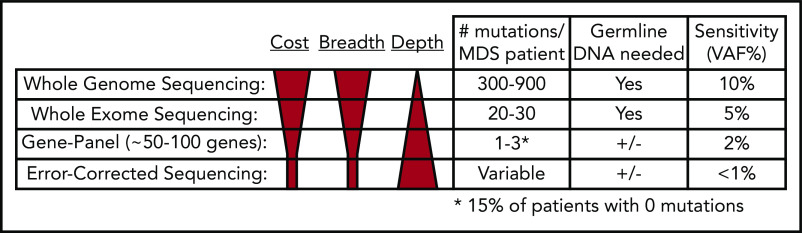
References
-
- Nimer SD. Myelodysplastic syndromes. Blood. 2008;111(10):4841-4851. - PubMed
-
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872-1885. - PubMed
-
- Swerdlow SH, Campo E, Harris NL, et al. . WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed Lyon, France: International Agency for Research on Cancer; 2008.
-
- Goldberg SL, Chen E, Corral M, et al. . Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J Clin Oncol. 2010;28(17):2847-2852. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
