

ERp29 as a regulator of Insulin biosynthesis
- PMID: 32433667
- PMCID: PMC7239452
- DOI: 10.1371/journal.pone.0233502
ERp29 as a regulator of Insulin biosynthesis
Erratum in
-
Correction: ERp29 as a regulator of Insulin biosynthesis.PLoS One. 2020 Aug 21;15(8):e0238264. doi: 10.1371/journal.pone.0238264. eCollection 2020. PLoS One. 2020. PMID: 32822440 Free PMC article.
Abstract
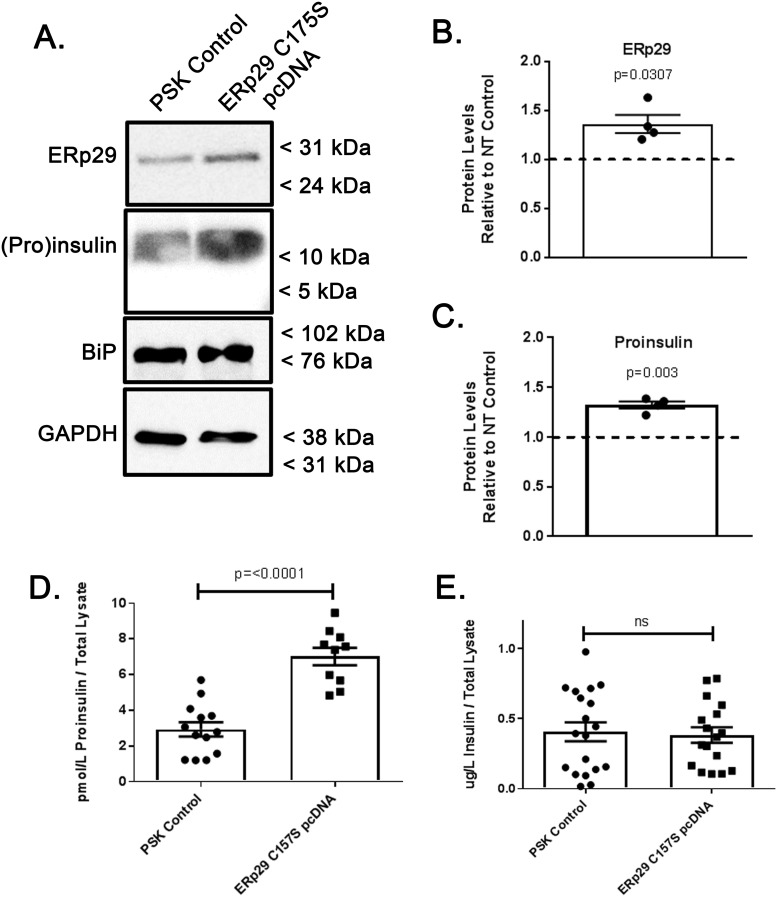
The environment within the Endoplasmic Reticulum (ER) influences Insulin biogenesis. In particular, ER stress may contribute to the development of Type 2 Diabetes (T2D) and Cystic Fibrosis Related Diabetes (CFRD), where evidence of impaired Insulin processing, including elevated secreted Proinsulin/Insulin ratios, are observed. Our group has established the role of a novel ER chaperone ERp29 (ER protein of 29 kDa) in the biogenesis of the Epithelial Sodium Channel, ENaC. The biogenesis of Insulin and ENaC share may key features, including their potential association with COP II machinery, their cleavage into a more active form in the Golgi or later compartments, and their ability to bypass such cleavage and remain in a less active form. Given these similarities we hypothesized that ERp29 is a critical factor in promoting the efficient conversion of Proinsulin to Insulin. Here, we confirmed that Proinsulin associates with the COP II vesicle cargo recognition component, Sec24D. When Sec24D expression was decreased, we observed a corresponding decrease in whole cell Proinsulin levels. In addition, we found that Sec24D associates with ERp29 in co-precipitation experiments and that ERp29 associates with Proinsulin in co-precipitation experiments. When ERp29 was overexpressed, a corresponding increase in whole cell Proinsulin levels was observed, while depletion of ERp29 decreased whole cell Proinsulin levels. Together, these data suggest a potential role for ERp29 in regulating Insulin biosynthesis, perhaps in promoting the exit of Proinsulin from the ER via Sec24D/COPII vesicles.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures






References
-
- Sundar Rajan S., et al., Endoplasmic reticulum (ER) stress & diabetes. Indian J Med Res, 2007. 125(3): p. 411–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
