

Integrated Structural Modeling of Full-Length LRH-1 Reveals Inter-domain Interactions Contribute to Receptor Structure and Function
- PMID: 32433991
- PMCID: PMC7347456
- DOI: 10.1016/j.str.2020.04.020
Integrated Structural Modeling of Full-Length LRH-1 Reveals Inter-domain Interactions Contribute to Receptor Structure and Function
Abstract
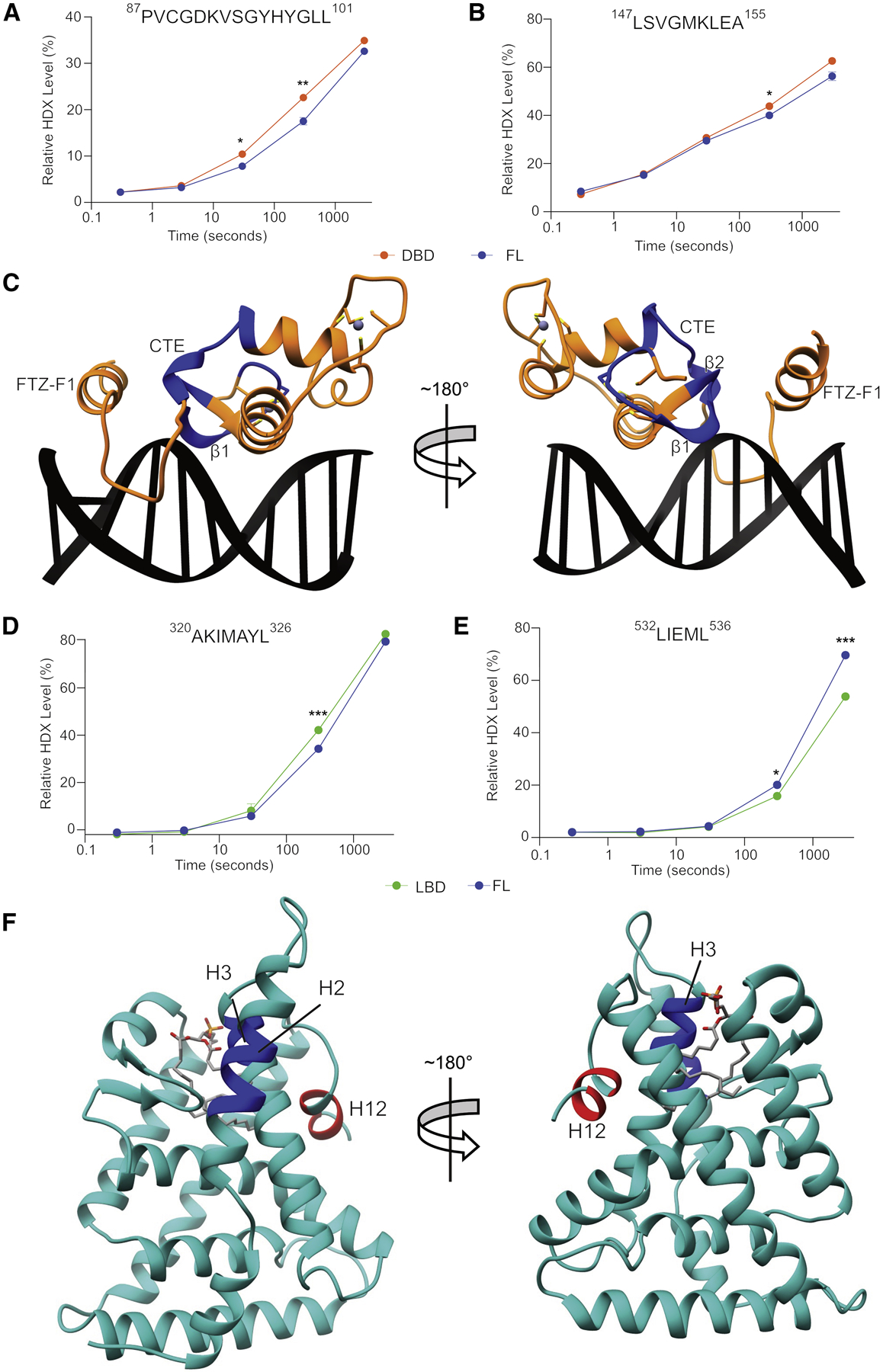
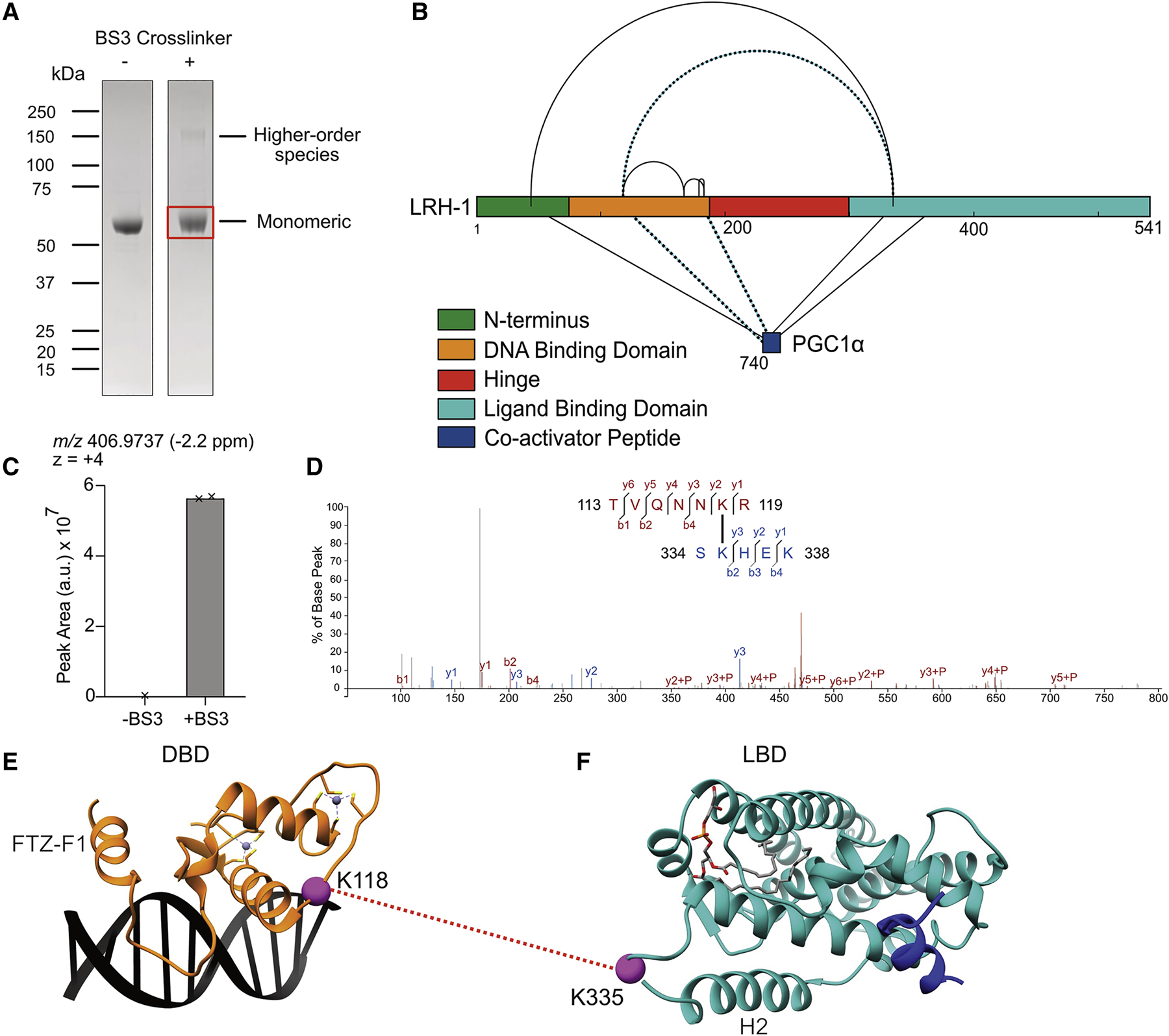
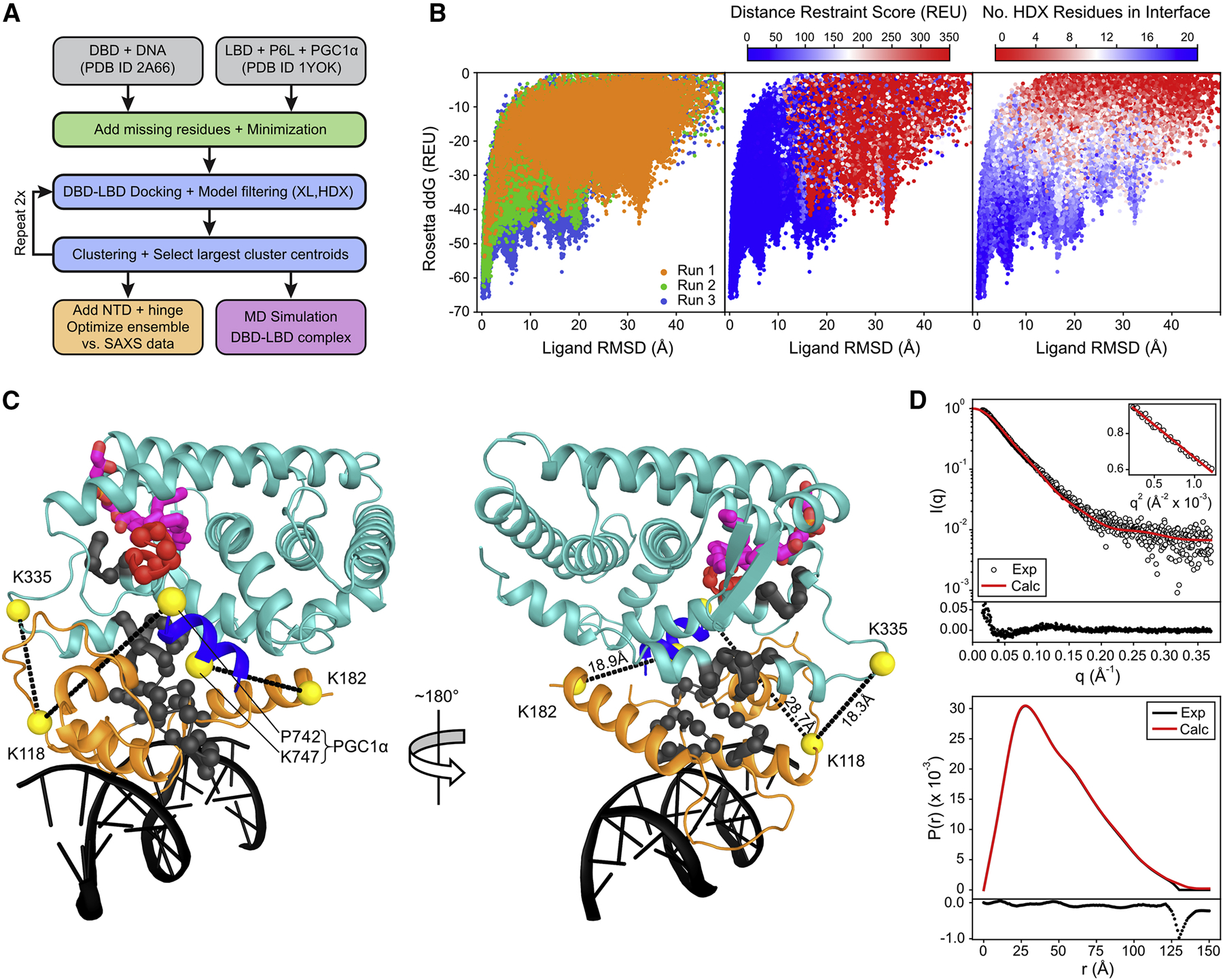
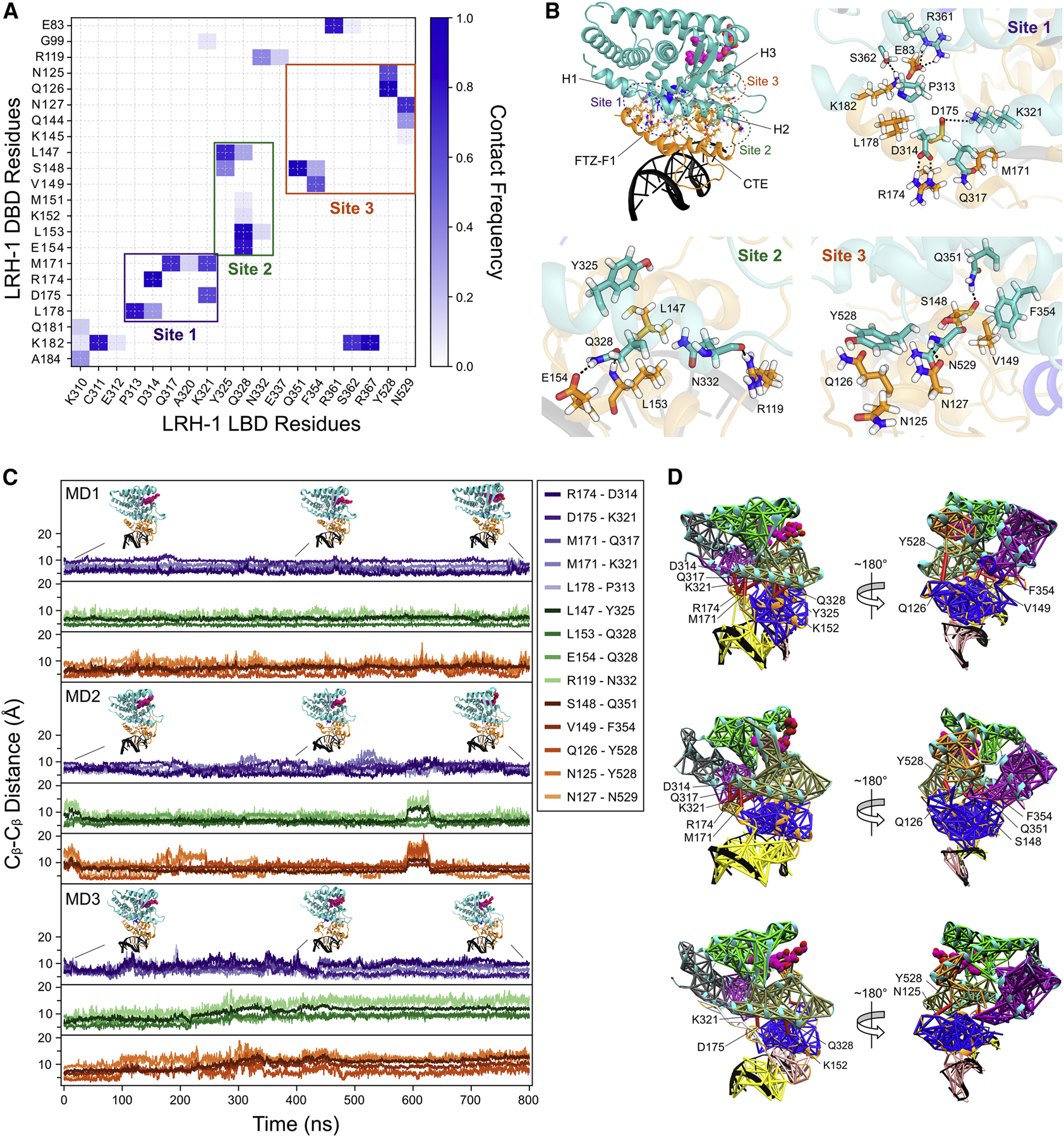
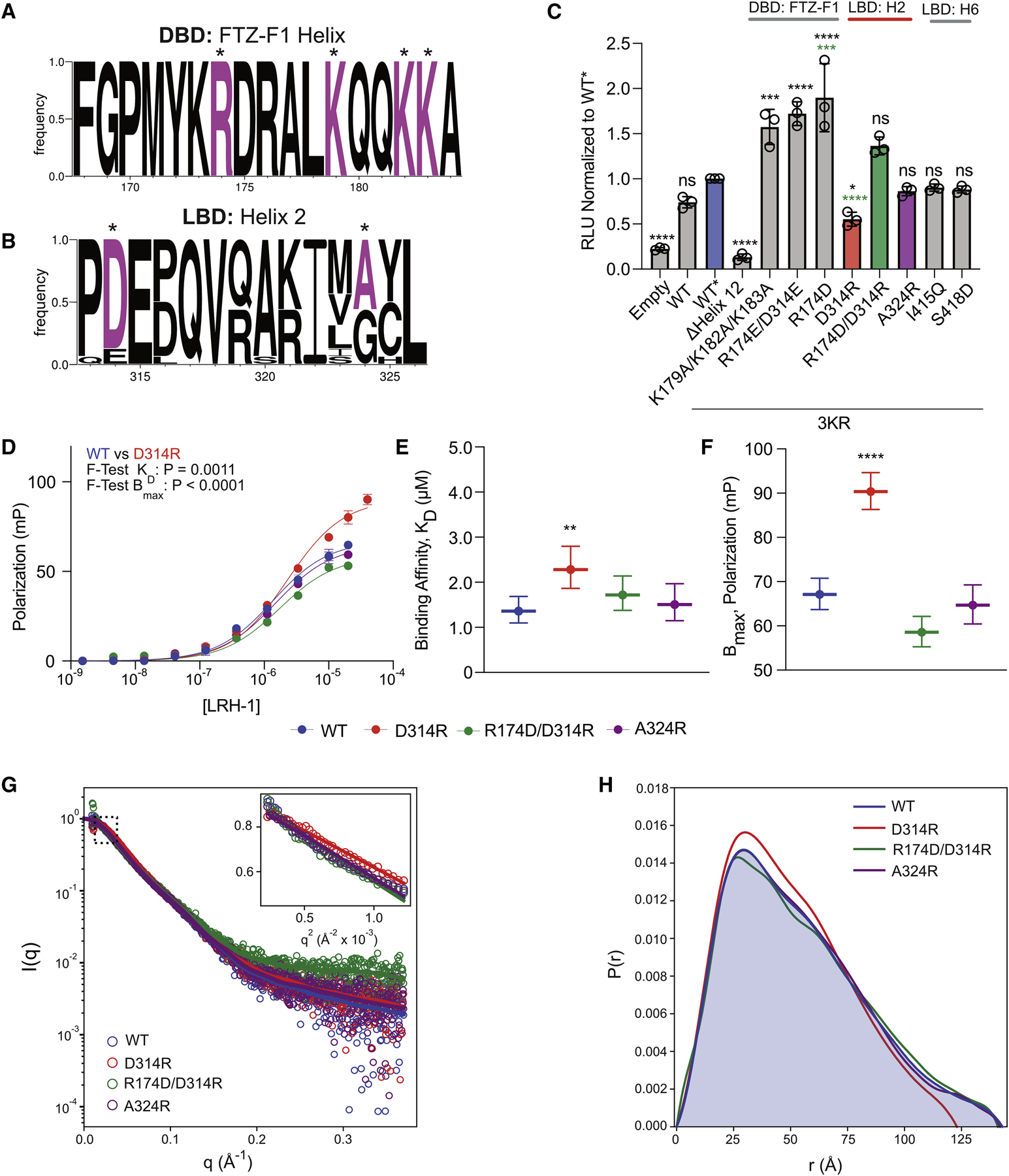
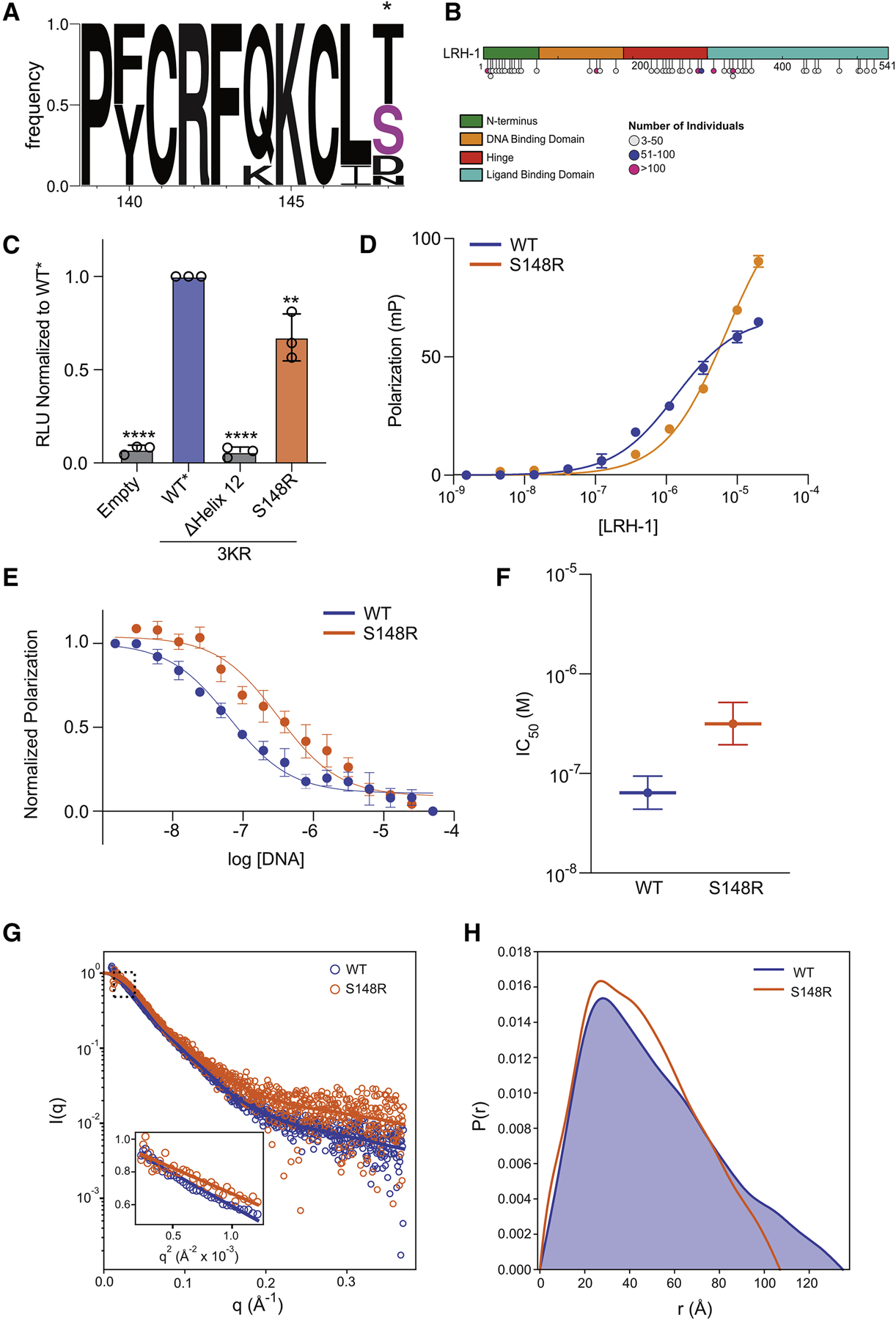
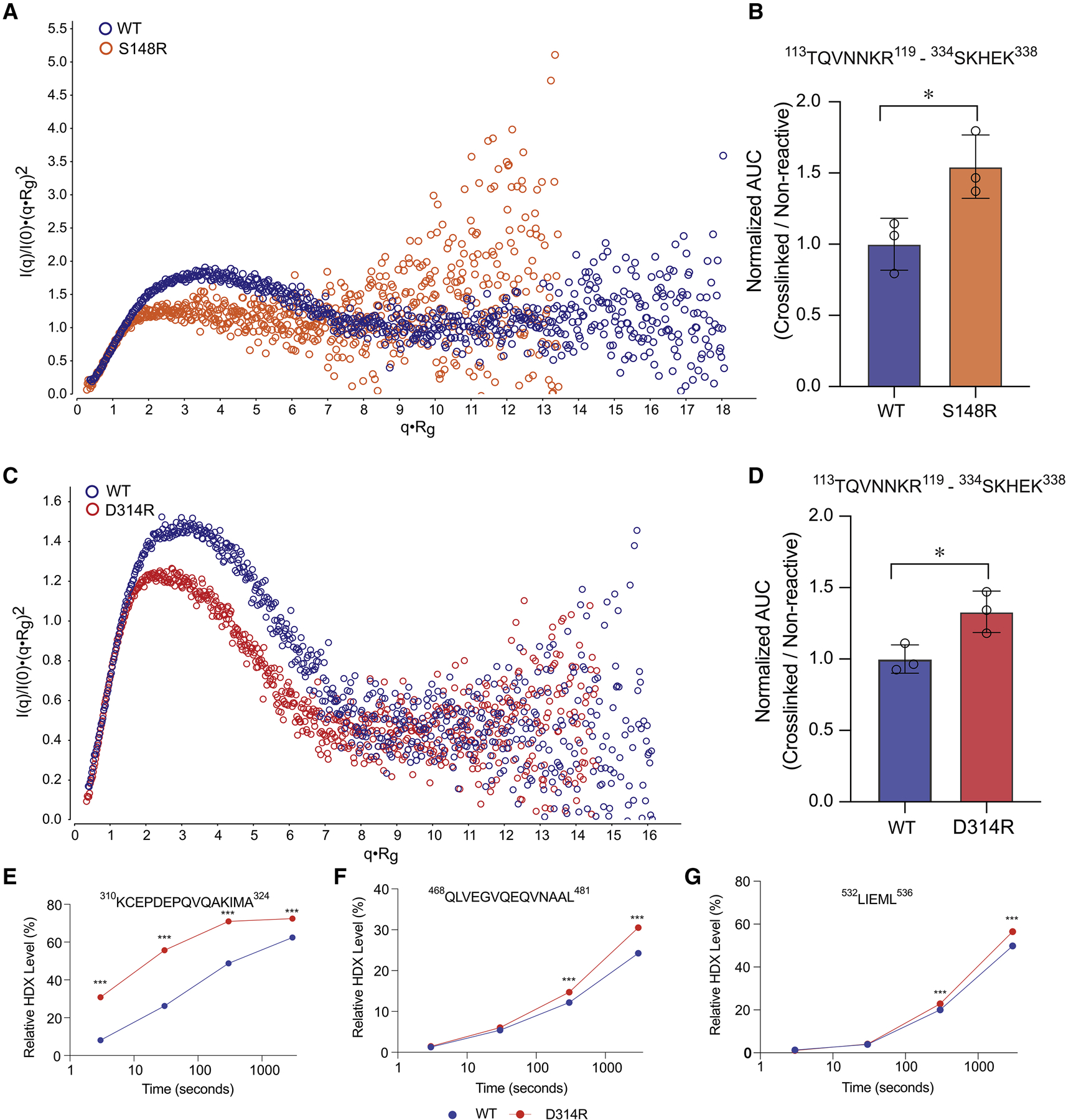
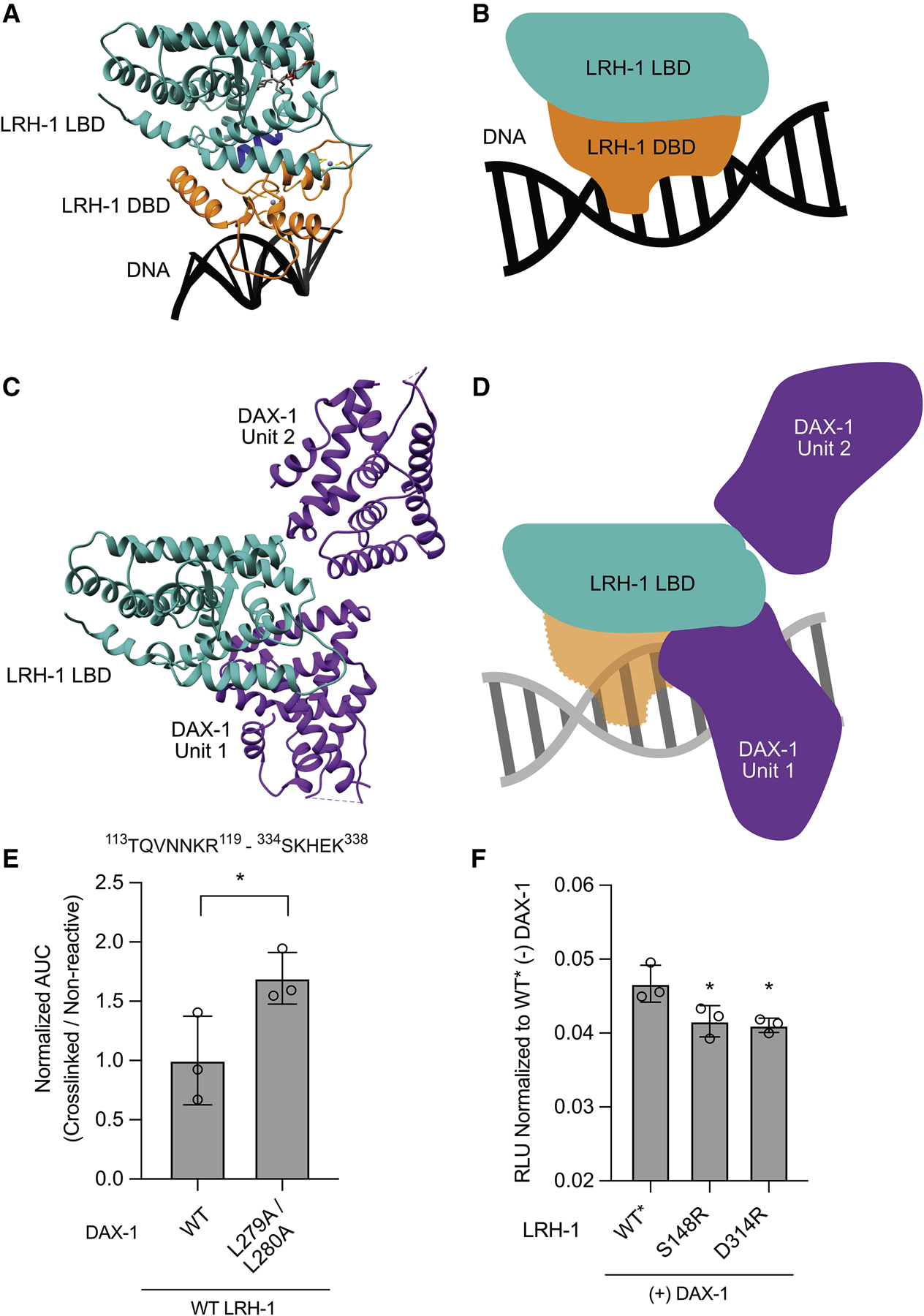
Liver receptor homolog-1 (LRH-1; NR5A2) is a nuclear receptor that regulates a diverse array of biological processes. In contrast to dimeric nuclear receptors, LRH-1 is an obligate monomer and contains a subtype-specific helix at the C terminus of the DNA-binding domain (DBD), termed FTZ-F1. Although detailed structural information is available for individual domains of LRH-1, it is unknown how these domains exist in the intact nuclear receptor. Here, we developed an integrated structural model of human full-length LRH-1 using a combination of HDX-MS, XL-MS, Rosetta computational docking, and SAXS. The model predicts the DBD FTZ-F1 helix directly interacts with ligand binding domain helix 2. We confirmed several other predicted inter-domain interactions via structural and functional analyses. Comparison between the LRH-1/Dax-1 co-crystal structure and the integrated model predicted and confirmed Dax-1 co-repressor to modulate LRH-1 inter-domain dynamics. Together, these data support individual LRH-1 domains interacting to influence receptor structure and function.
Keywords: BS3; Dax1 Dax-1 Nr0b1; benzophenone artificial amino acid; disulfide crosslink mass spectrometry; hydrogen-deuterium exchange mass spectrometry; integrated structural modeling; nuclear lipids; nuclear phospholipid signaling; nuclear receptor lipidomics; small-angle X-ray scattering; β-catenin CTNNB1.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
Crystal structure of the human LRH-1 DBD-DNA complex reveals Ftz-F1 domain positioning is required for receptor activity.J Mol Biol. 2005 Dec 16;354(5):1091-102. doi: 10.1016/j.jmb.2005.10.009. Epub 2005 Oct 27. J Mol Biol. 2005. PMID: 16289203
-
Structure and Dynamics of the Liver Receptor Homolog 1-PGC1α Complex.Mol Pharmacol. 2017 Jul;92(1):1-11. doi: 10.1124/mol.117.108514. Epub 2017 Mar 31. Mol Pharmacol. 2017. PMID: 28363985 Free PMC article.
-
Structure of Liver Receptor Homolog-1 (NR5A2) with PIP3 hormone bound in the ligand binding pocket.J Struct Biol. 2015 Dec;192(3):342-348. doi: 10.1016/j.jsb.2015.09.012. Epub 2015 Sep 28. J Struct Biol. 2015. PMID: 26416531 Free PMC article.
-
LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis.Trends Cell Biol. 2004 May;14(5):250-60. doi: 10.1016/j.tcb.2004.03.008. Trends Cell Biol. 2004. PMID: 15130581 Review.
-
The Orphan Nuclear Receptors Steroidogenic Factor-1 and Liver Receptor Homolog-1: Structure, Regulation, and Essential Roles in Mammalian Reproduction.Physiol Rev. 2019 Apr 1;99(2):1249-1279. doi: 10.1152/physrev.00019.2018. Physiol Rev. 2019. PMID: 30810078 Review.
Cited by
-
New High-Throughput Screen Discovers Novel Ligands of Full-Length Nuclear Receptor LRH-1.ACS Chem Biol. 2023 May 19;18(5):1101-1114. doi: 10.1021/acschembio.2c00805. Epub 2023 Apr 19. ACS Chem Biol. 2023. PMID: 37074920 Free PMC article.
-
Differential Modulation of Nuclear Receptor LRH-1 through Targeting Buried and Surface Regions of the Binding Pocket.J Med Chem. 2022 May 12;65(9):6888-6902. doi: 10.1021/acs.jmedchem.2c00235. Epub 2022 May 3. J Med Chem. 2022. PMID: 35503419 Free PMC article.
-
SF-1 Induces Nuclear PIP2.Biomolecules. 2023 Oct 12;13(10):1509. doi: 10.3390/biom13101509. Biomolecules. 2023. PMID: 37892191 Free PMC article.
-
NR5A2 promotes malignancy progression and mediates the effect of cisplatin in cutaneous squamous cell carcinoma.Immun Inflamm Dis. 2024 Feb;12(2):e1172. doi: 10.1002/iid3.1172. Immun Inflamm Dis. 2024. PMID: 38358044 Free PMC article.
-
Conformational Changes of RORγ During Response Element Recognition and Coregulator Engagement.J Mol Biol. 2021 Nov 5;433(22):167258. doi: 10.1016/j.jmb.2021.167258. Epub 2021 Sep 20. J Mol Biol. 2021. PMID: 34547329 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- T32 GM007628/GM/NIGMS NIH HHS/United States
- P30 DK063720/DK/NIDDK NIH HHS/United States
- R01 GM080403/GM/NIGMS NIH HHS/United States
- P30 GM124169/GM/NIGMS NIH HHS/United States
- K01 CA172957/CA/NCI NIH HHS/United States
- S10 OD018483/OD/NIH HHS/United States
- F31 DK120214/DK/NIDDK NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
- S10 OD023680/OD/NIH HHS/United States
- R21 CA243036/CA/NCI NIH HHS/United States
- R01 GM132592/GM/NIGMS NIH HHS/United States
- T32 DK007563/DK/NIDDK NIH HHS/United States
- P60 DK020593/DK/NIDDK NIH HHS/United States
- R01 GM138873/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
