

To Dereplicate or Not To Dereplicate?
- PMID: 32434845
- PMCID: PMC7380574
- DOI: 10.1128/mSphere.00971-19
To Dereplicate or Not To Dereplicate?
Abstract
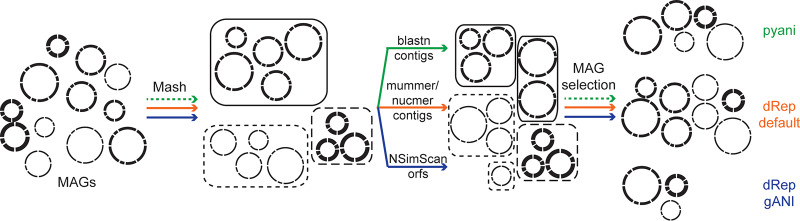
Metagenome-assembled genomes (MAGs) expand our understanding of microbial diversity, evolution, and ecology. Concerns have been raised on how sequencing, assembly, binning, and quality assessment tools may result in MAGs that do not reflect single populations in nature. Here, we reflect on another issue, i.e., how to handle highly similar MAGs assembled from independent data sets. Obtaining multiple genomic representatives for a species is highly valuable, as it allows for population genomic analyses; however, when retaining genomes of closely related populations, it complicates MAG quality assessment and abundance inferences. We show that (i) published data sets contain a large fraction of MAGs sharing >99% average nucleotide identity, (ii) different software packages and parameters used to resolve this redundancy remove very different numbers of MAGs, and (iii) the removal of closely related genomes leads to losses of population-specific auxiliary genes. Finally, we highlight some approaches that can infer strain-specific dynamics across a sample series without dereplication.
Keywords: MAG; binning; dereplication; metagenomics; population genomics; software.
Copyright © 2020 Evans and Denef.
Figures

References
-
- Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF. 2016. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 7:13219. doi: 10.1038/ncomms13219. - DOI - PMC - PubMed
-
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N. 2019. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176:649–662. doi: 10.1016/j.cell.2019.01.001. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
