

doi: 10.1021/acs.jctc.0c00338.
Epub 2020 Jun 12.
Pseudo-Improper-Dihedral Model for Intrinsically Disordered Proteins
Affiliations
- PMID: 32436706
- PMCID: PMC7588027
- DOI: 10.1021/acs.jctc.0c00338
Item in Clipboard
Pseudo-Improper-Dihedral Model for Intrinsically Disordered Proteins
J Chem Theory Comput.
.
Abstract
We present a new coarse-grained Cα-based protein model with a nonradial multibody pseudo-improper-dihedral potential that is transferable, time-independent, and suitable for molecular dynamics. It captures the nature of backbone and side-chain interactions between amino acid residues by adapting a simple improper dihedral term for a one-bead-per-residue model. It is parameterized for intrinsically disordered proteins and applicable to simulations of such proteins and their assemblies on millisecond time scales.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
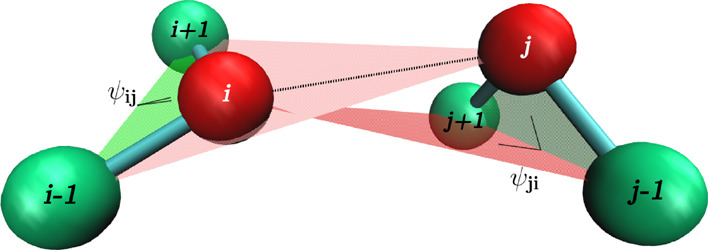
Idea of the PID angles.
The interaction between residues i and j involves angles Ψij (defined by i – 1, i, i + 1,
and j beads)
and Ψji (defined by j – 1, j, j + 1, and i beads).
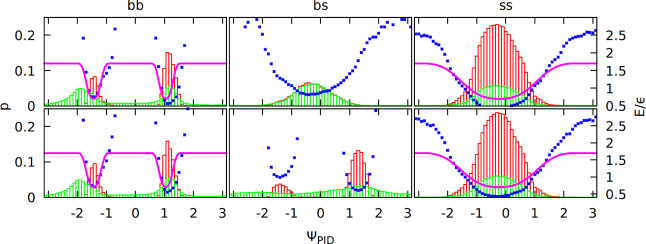
Distributions
of the PID angles in the contacts from the PDB survey
that are only of one type (green histograms). Local i, i + 3 and i, i + 4 contacts are excluded. Each contact has two angles. Distribution
of the first (ΨPIDij) is on the top panels, and that of the
second (ΨPIDji) is on the bottom panels. Subdistributions
made from contacts that obey the directional criteria defined in ref (37) are shown as red histograms.
The potential resulting from the Boltzmann inversion procedure (blue
dots; unit of energy, ε ≈ 1.5 kcal/mol) was fitted to
an analytical function (purple line).
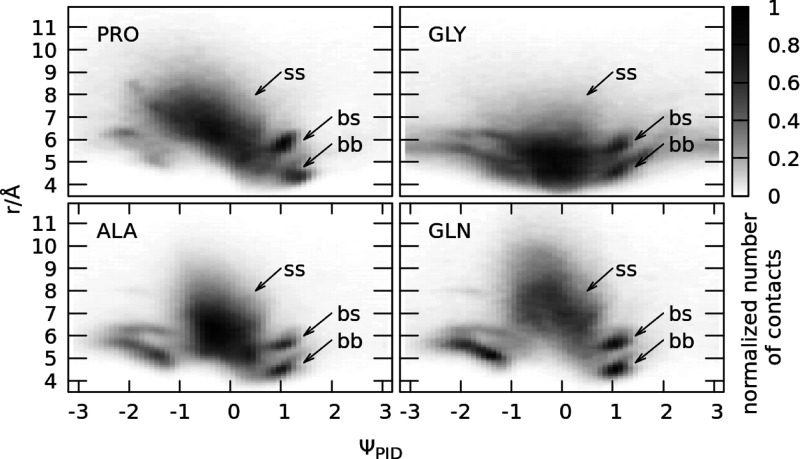
Two-dimensional distributions
of contacts, where the PID angle
ΨPID is on one axis, and the Cα–Cα distance r is on the other axis. The i, i + 3 and i, i + 4 contacts are excluded. The distributions include each
contact obtained in the PDB survey where at least one residue in the
pair was of the given amino acid type (PRO, GLY, ALA, and GLN). The
PID angle ΨPID for each contact is the one associated
with this residue (if both residues were the same amino acid, then
it corresponds to two counts with two different PID angles).
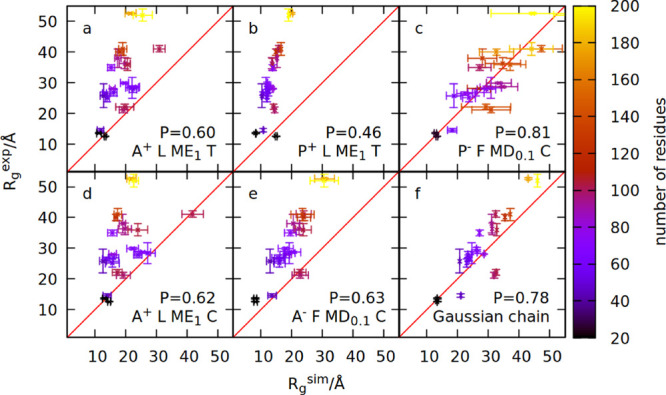
Comparison between the simulated and measured radius of gyration
for 23 IDPs. Panels (a)–(f) show simulation results obtained
within six different models. The model names are specified in the
panels and defined in the text. The value of the Pearson coefficient
is also indicated in each panel. Colors indicate the number of residues
constituting a given IDP.
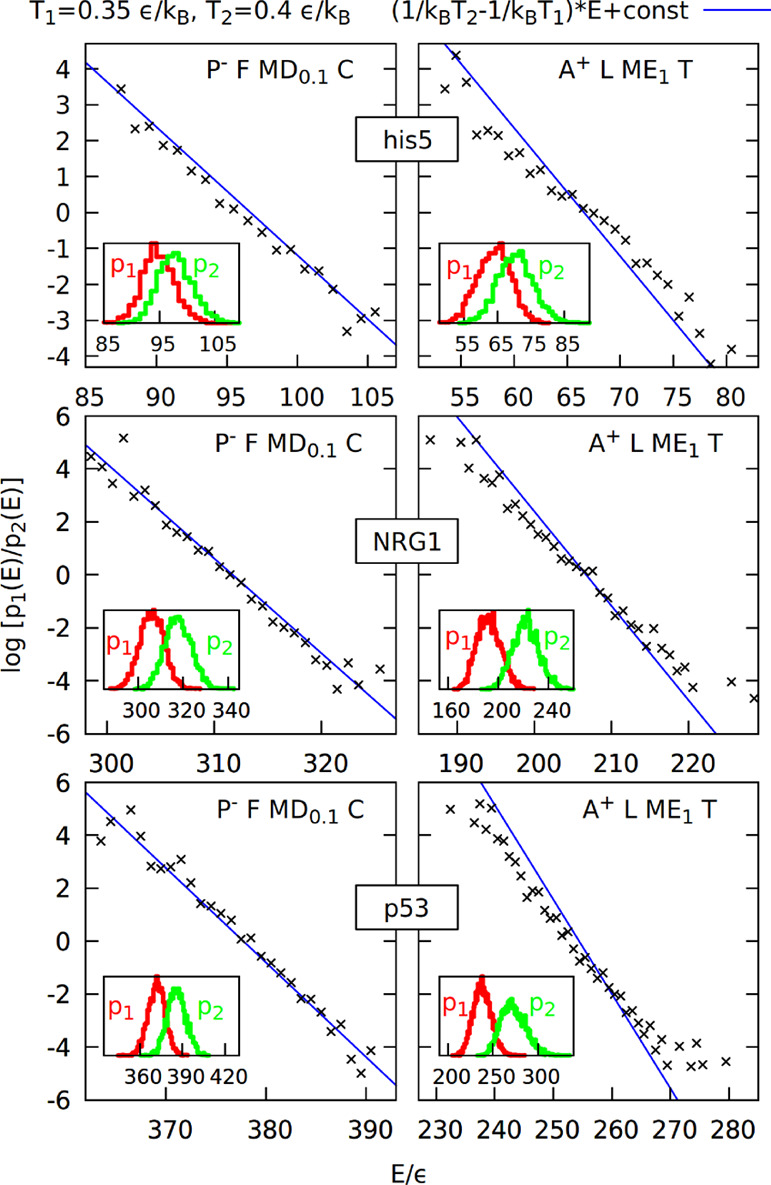
Histogram test for the new model (P– F MD0.1 C) and previous model (A+ L ME1 T)
based on simulations of proteins his5, NRG1,
and p53 at temperatures 0.35 and 0.4 ε/kB. Insets show the energy histograms binned every 1 ε
and used to construct the quantity log(p1(E)/p2(E)) shown on the main plots as a function of energy.
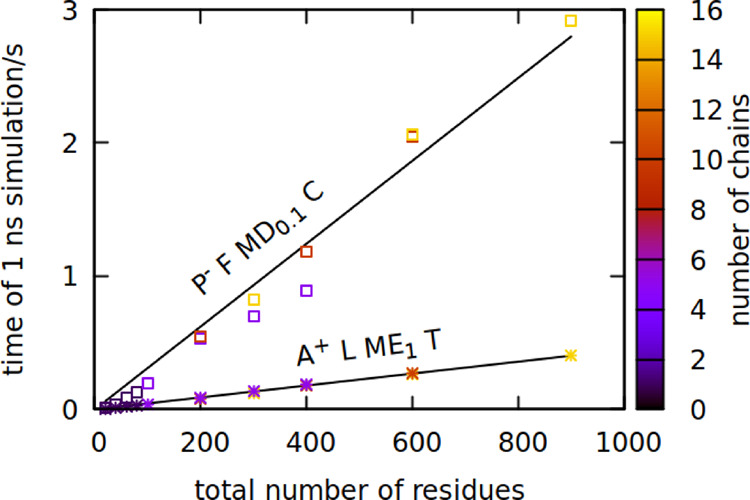
Physical time of 1 ns-simulation
run on a single core for the new
model (P– F MD0.1 C, squares) and previous
model (A+ L ME1 T, stars) as a function of the
system size. The number of protein chains in the simulation is indicated
by color. The system density of multichain simulations was set to
1 residue/nm3. Fitted lines have coefficients of 0.003
and 0.0005 s/residue, respectively, with a fit error in the order
of 0.0001 s/residue.
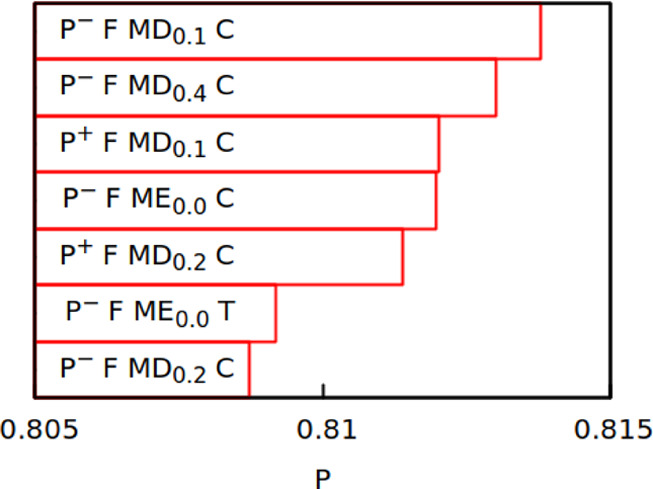
Top seven variants of
the PID model ranked by their Pearson coefficient.
References
-
- Bixon M.; Lifson S. Potential functions and conformations in cycloalkanes. Tetrahedron 1967, 23, 769–784. 10.1016/0040-4020(67)85023-3. - DOI
-
- Lifson S.; Warshel A. Consistent force field for calculations of conformations, vibrational spectra, and enthalpies of cycloalkane and n-alkane molecules. J. Chem. Phys. 1968, 49, 5116–5129. 10.1063/1.1670007. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources