

FoxO1-Dio2 signaling axis governs cardiomyocyte thyroid hormone metabolism and hypertrophic growth
- PMID: 32439985
- PMCID: PMC7242347
- DOI: 10.1038/s41467-020-16345-y
FoxO1-Dio2 signaling axis governs cardiomyocyte thyroid hormone metabolism and hypertrophic growth
Abstract
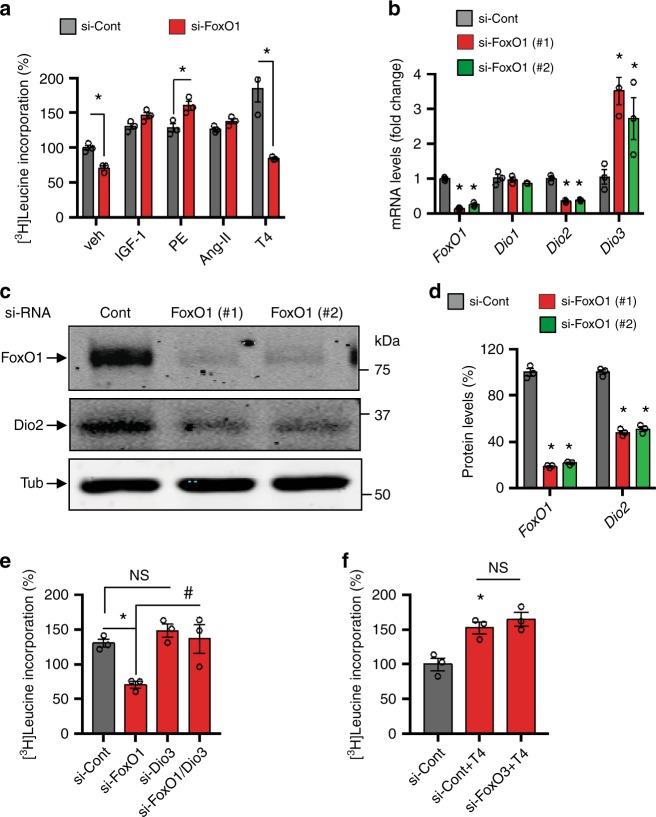
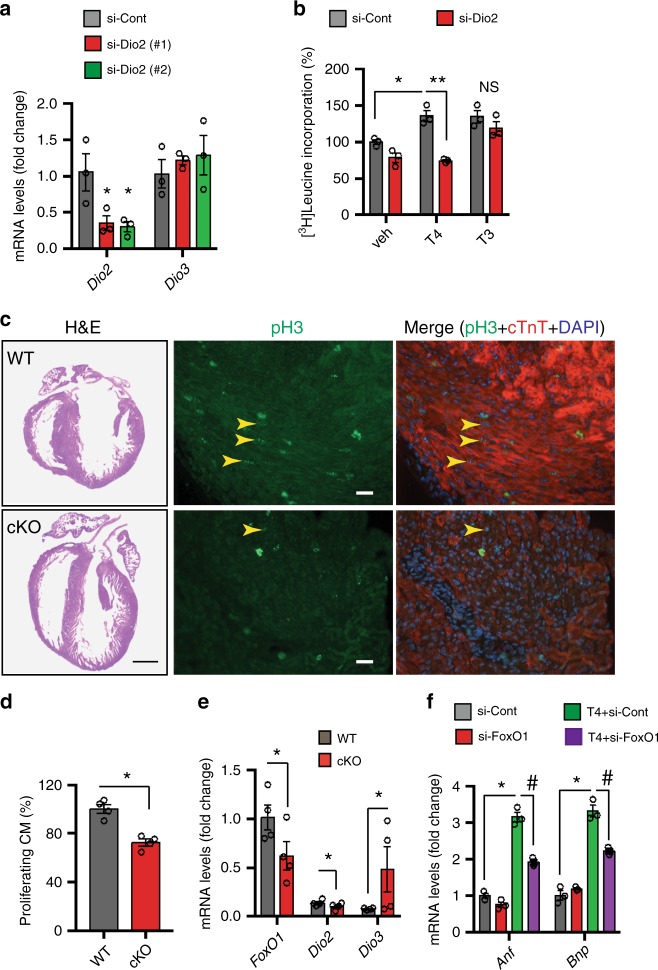
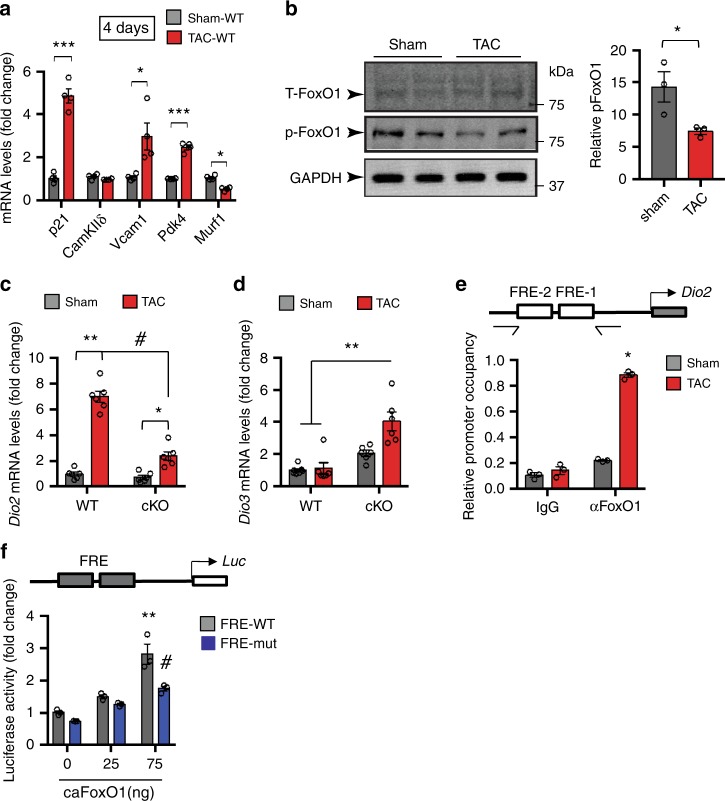
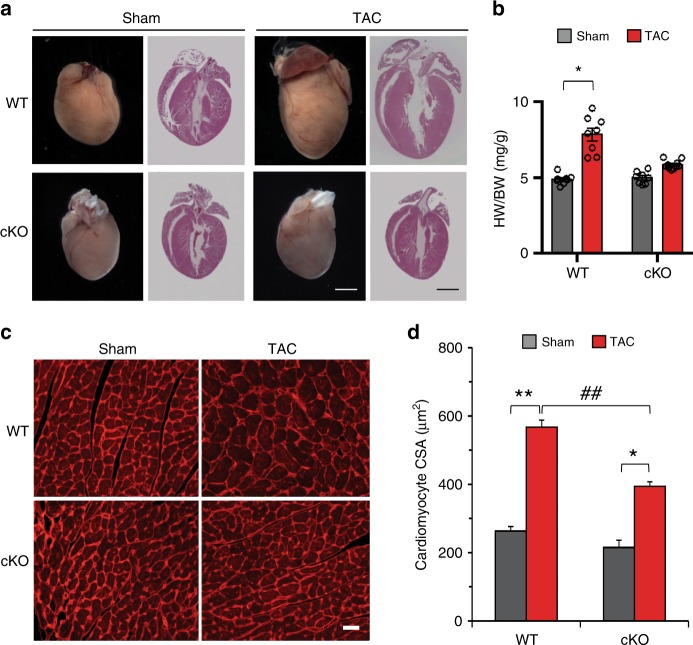
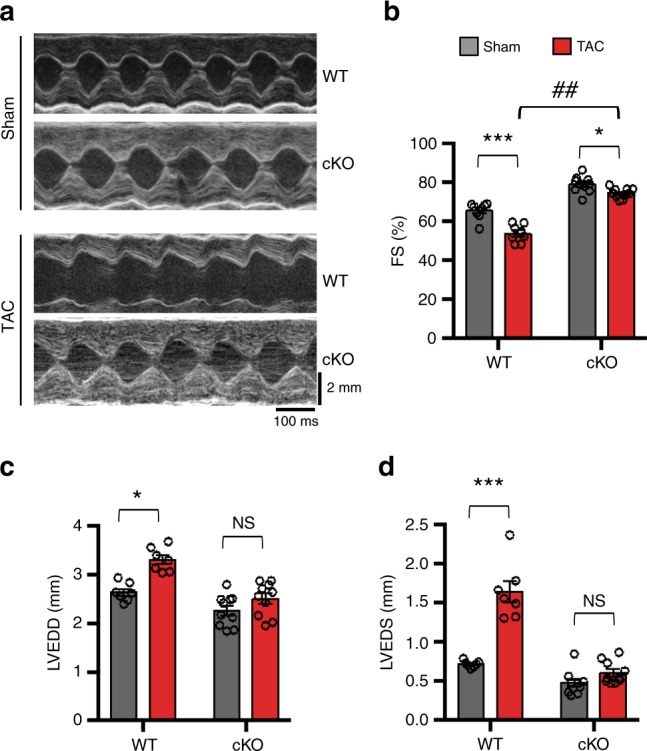
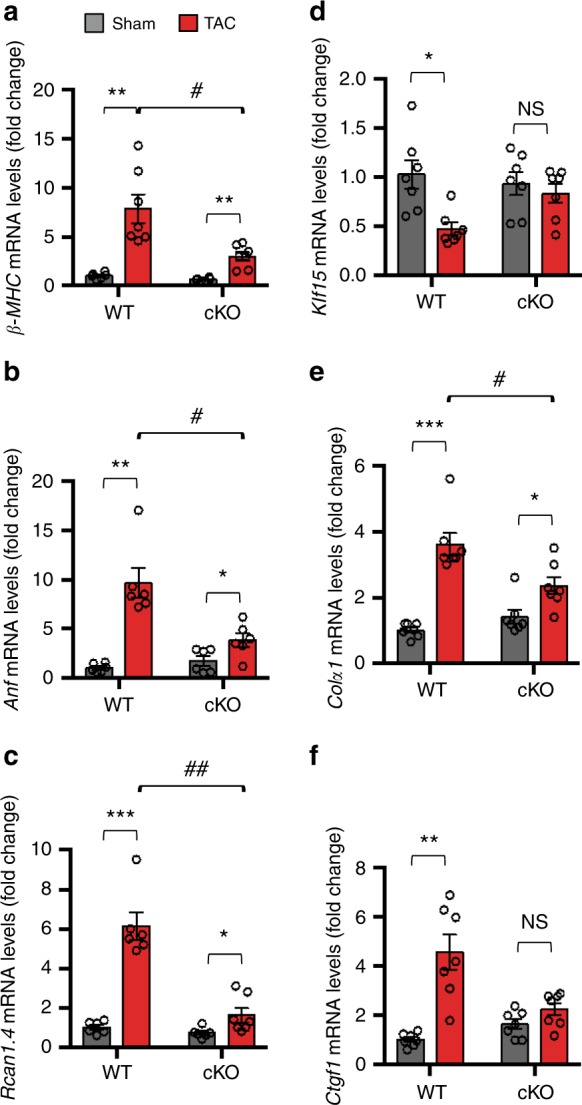
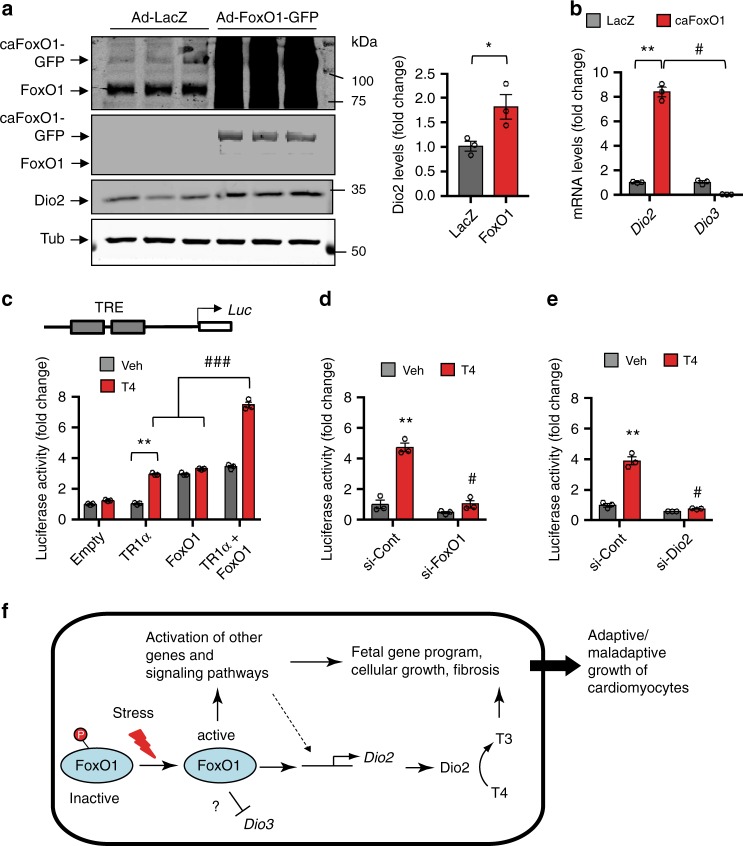
Forkhead box O (FoxO) proteins and thyroid hormone (TH) have well established roles in cardiovascular morphogenesis and remodeling. However, specific role(s) of individual FoxO family members in stress-induced growth and remodeling of cardiomyocytes remains unknown. Here, we report that FoxO1, but not FoxO3, activity is essential for reciprocal regulation of types II and III iodothyronine deiodinases (Dio2 and Dio3, respectively), key enzymes involved in intracellular TH metabolism. We further show that Dio2 is a direct transcriptional target of FoxO1, and the FoxO1-Dio2 axis governs TH-induced hypertrophic growth of neonatal cardiomyocytes in vitro and in vivo. Utilizing transverse aortic constriction as a model of hemodynamic stress in wild-type and cardiomyocyte-restricted FoxO1 knockout mice, we unveil an essential role for the FoxO1-Dio2 axis in afterload-induced pathological cardiac remodeling and activation of TRα1. These findings demonstrate a previously unrecognized FoxO1-Dio2 signaling axis in stress-induced cardiomyocyte growth and remodeling and intracellular TH homeostasis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Benjamin EJ, et al. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. - PubMed
-
- Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327–334. - PubMed
-
- Hill JA. Braking bad hypertrophy. N. Engl. J. Med. 2015;372:2160–2162. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
