

GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins
- PMID: 32440658
- PMCID: PMC7222226
- DOI: 10.1093/nargab/lqaa026
GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins
Abstract
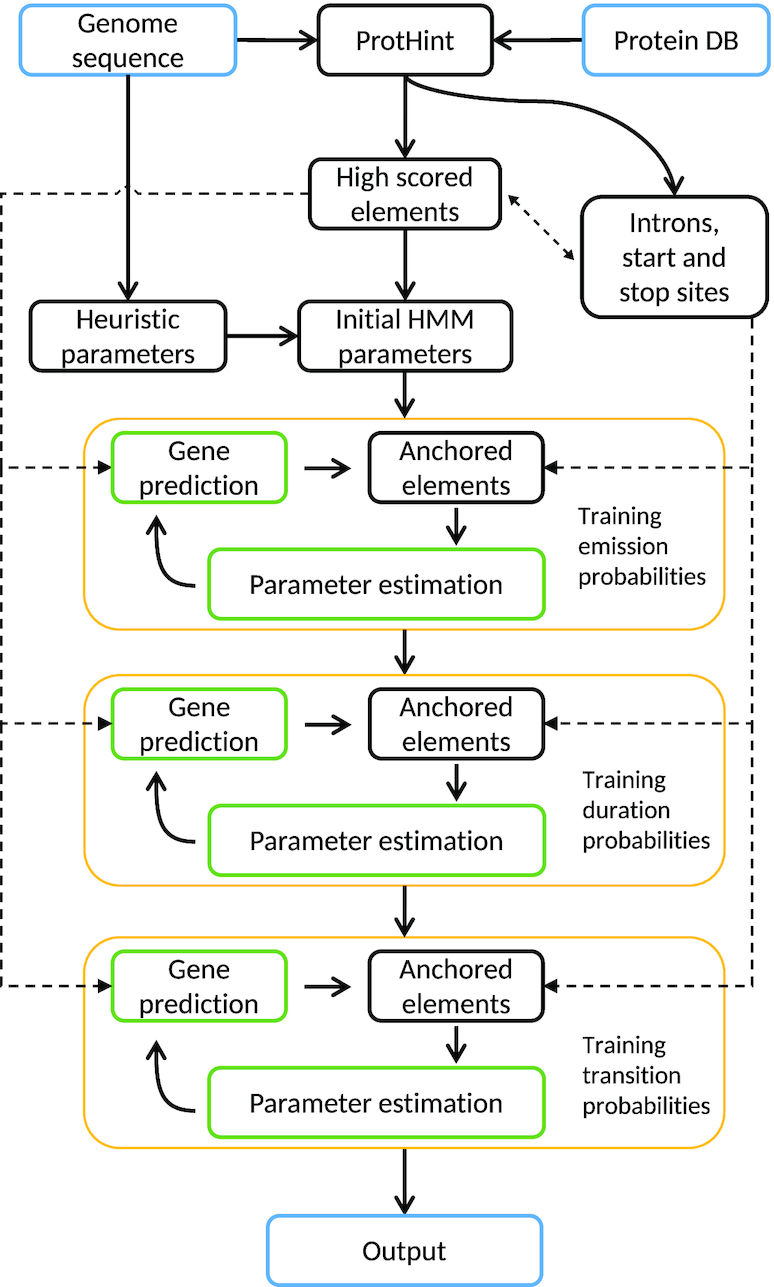
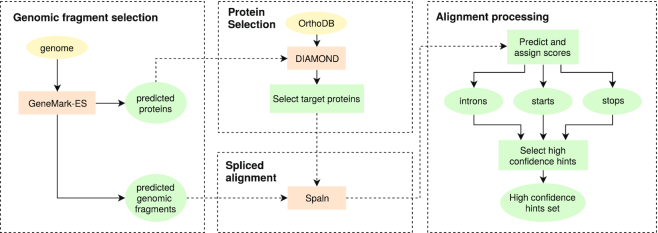
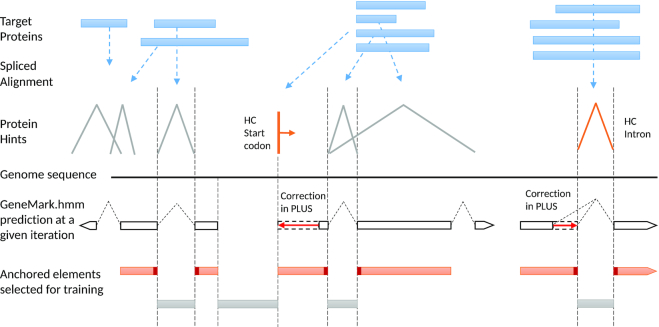
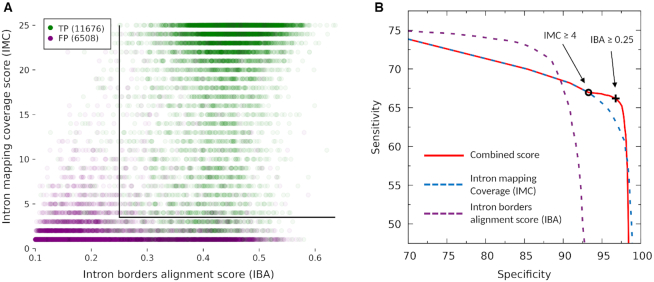
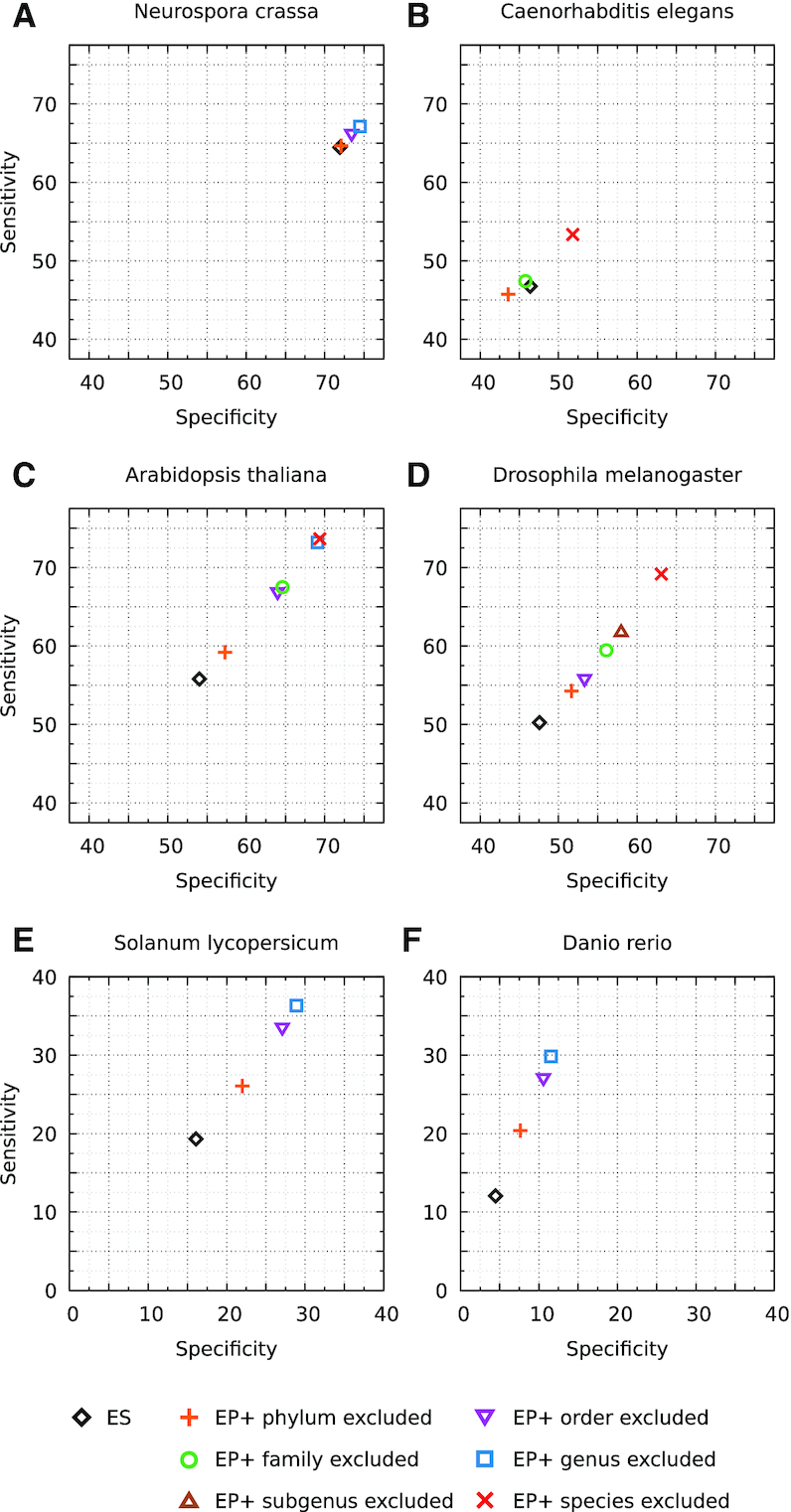
We have made several steps toward creating a fast and accurate algorithm for gene prediction in eukaryotic genomes. First, we introduced an automated method for efficient ab initio gene finding, GeneMark-ES, with parameters trained in iterative unsupervised mode. Next, in GeneMark-ET we proposed a method of integration of unsupervised training with information on intron positions revealed by mapping short RNA reads. Now we describe GeneMark-EP, a tool that utilizes another source of external information, a protein database, readily available prior to the start of a sequencing project. A new specialized pipeline, ProtHint, initiates massive protein mapping to genome and extracts hints to splice sites and translation start and stop sites of potential genes. GeneMark-EP uses the hints to improve estimation of model parameters as well as to adjust coordinates of predicted genes if they disagree with the most reliable hints (the -EP+ mode). Tests of GeneMark-EP and -EP+ demonstrated improvements in gene prediction accuracy in comparison with GeneMark-ES, while the GeneMark-EP+ showed higher accuracy than GeneMark-ET. We have observed that the most pronounced improvements in gene prediction accuracy happened in large eukaryotic genomes.
© The Author(s) 2019. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
-
- Hoff K.J., Stanke M.. Predicting genes in single genomes with AUGUSTUS. Curr. Protoc. Bioinformatics. 2019; 65:e57. - PubMed
-
- Foissac S., Gouzy J., Rombauts S., Mathe C., Amselem J., Sterck L., Van de Peer Y., Rouze P., Schiex T.. Genome annotation in plants and fungi: EuGene as a model platform. Curr. Bioinformatics. 2008; 3:87–97.
-
- Sallet E., Gouzy J., Schiex T.. EuGene: an automated integrative gene finder for eukaryotes and prokaryotes. Methods Mol. Biol. 2019; 1962:97–120. - PubMed
-
- Behr J., Bohnert R., Zeller G., Schweikert G., Hartmann L., Rätsch G.. Next generation genome annotation with mGene.ngs. BMC Bioinformatics. 2010; 11:O8.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
