

Antifungal Drug Resistance: Molecular Mechanisms in Candida albicans and Beyond
- PMID: 32441527
- PMCID: PMC8519031
- DOI: 10.1021/acs.chemrev.0c00199
Antifungal Drug Resistance: Molecular Mechanisms in Candida albicans and Beyond
Abstract
Fungal infections are a major contributor to infectious disease-related deaths across the globe. Candida species are among the most common causes of invasive mycotic disease, with Candida albicans reigning as the leading cause of invasive candidiasis. Given that fungi are eukaryotes like their human host, the number of unique molecular targets that can be exploited for antifungal development remains limited. Currently, there are only three major classes of drugs approved for the treatment of invasive mycoses, and the efficacy of these agents is compromised by the development of drug resistance in pathogen populations. Notably, the emergence of additional drug-resistant species, such as Candida auris and Candida glabrata, further threatens the limited armamentarium of antifungals available to treat these serious infections. Here, we describe our current arsenal of antifungals and elaborate on the resistance mechanisms Candida species possess that render them recalcitrant to therapeutic intervention. Finally, we highlight some of the most promising therapeutic strategies that may help combat antifungal resistance, including combination therapy, targeting fungal-virulence traits, and modulating host immunity. Overall, a thorough understanding of the mechanistic principles governing antifungal drug resistance is fundamental for the development of novel therapeutics to combat current and emerging fungal threats.
Conflict of interest statement
The authors declare the following competing financial interest(s): L.E.C. is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics. L.E.C. is a consultant for Boragen, a small-molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health.
Figures
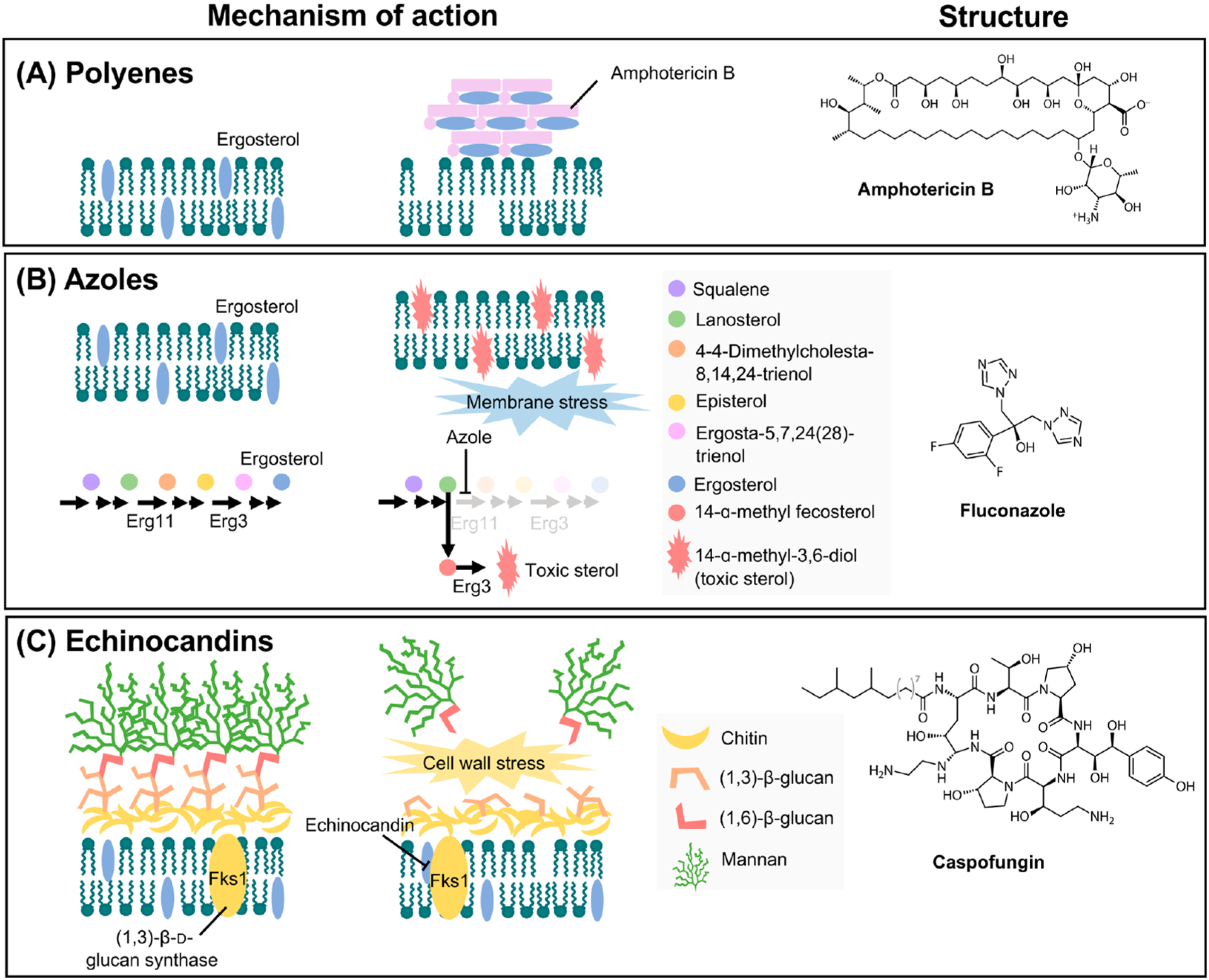
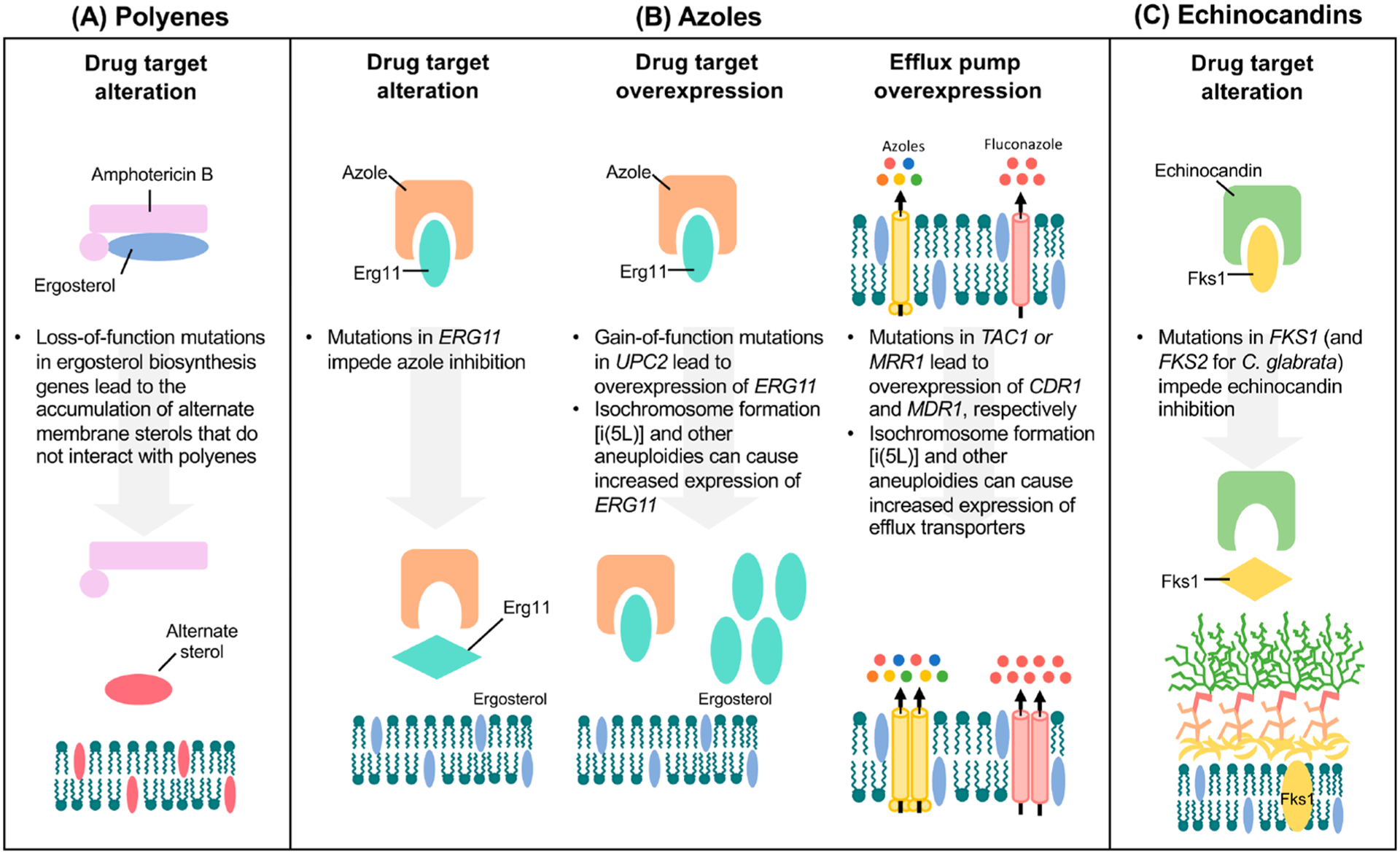
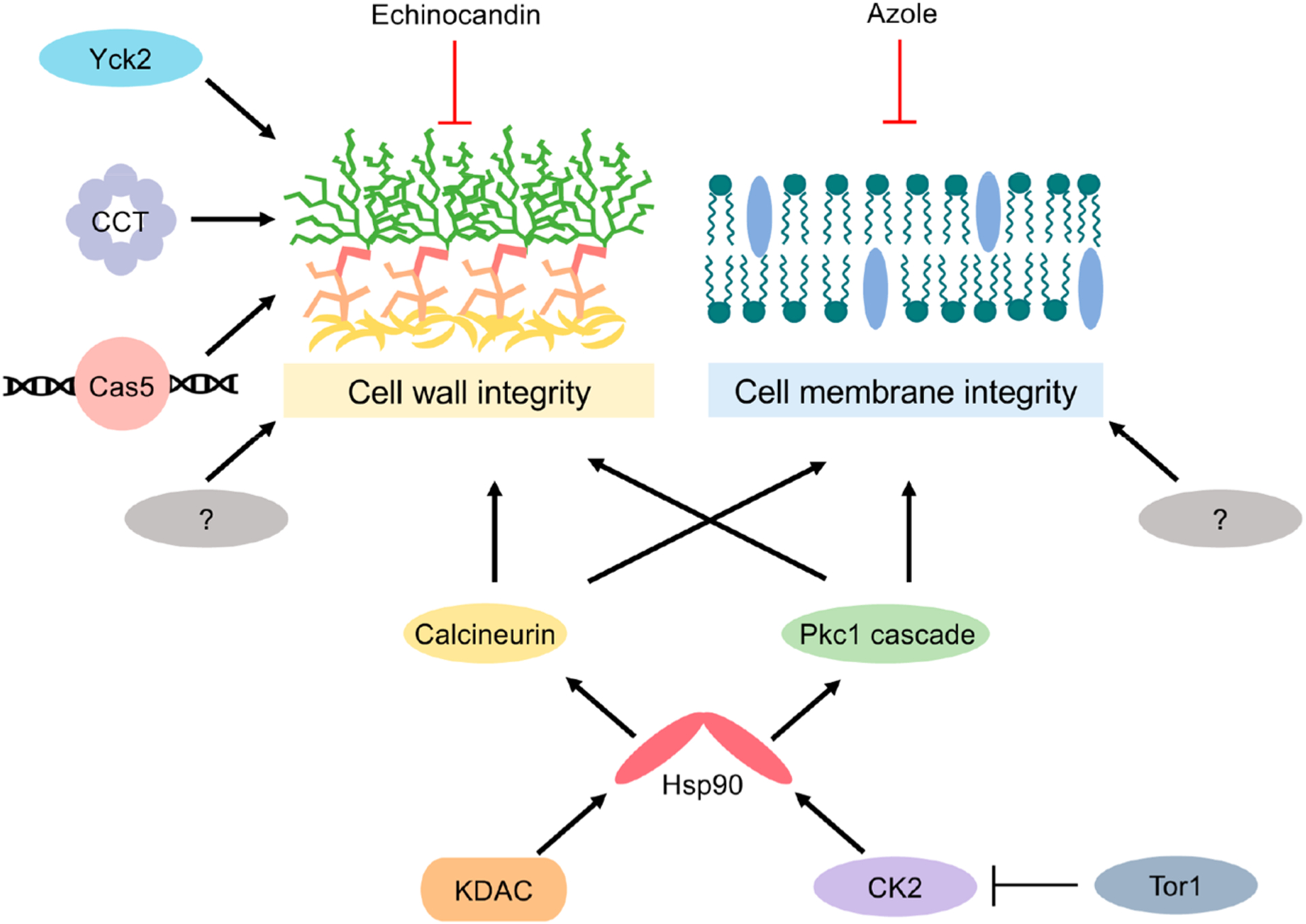
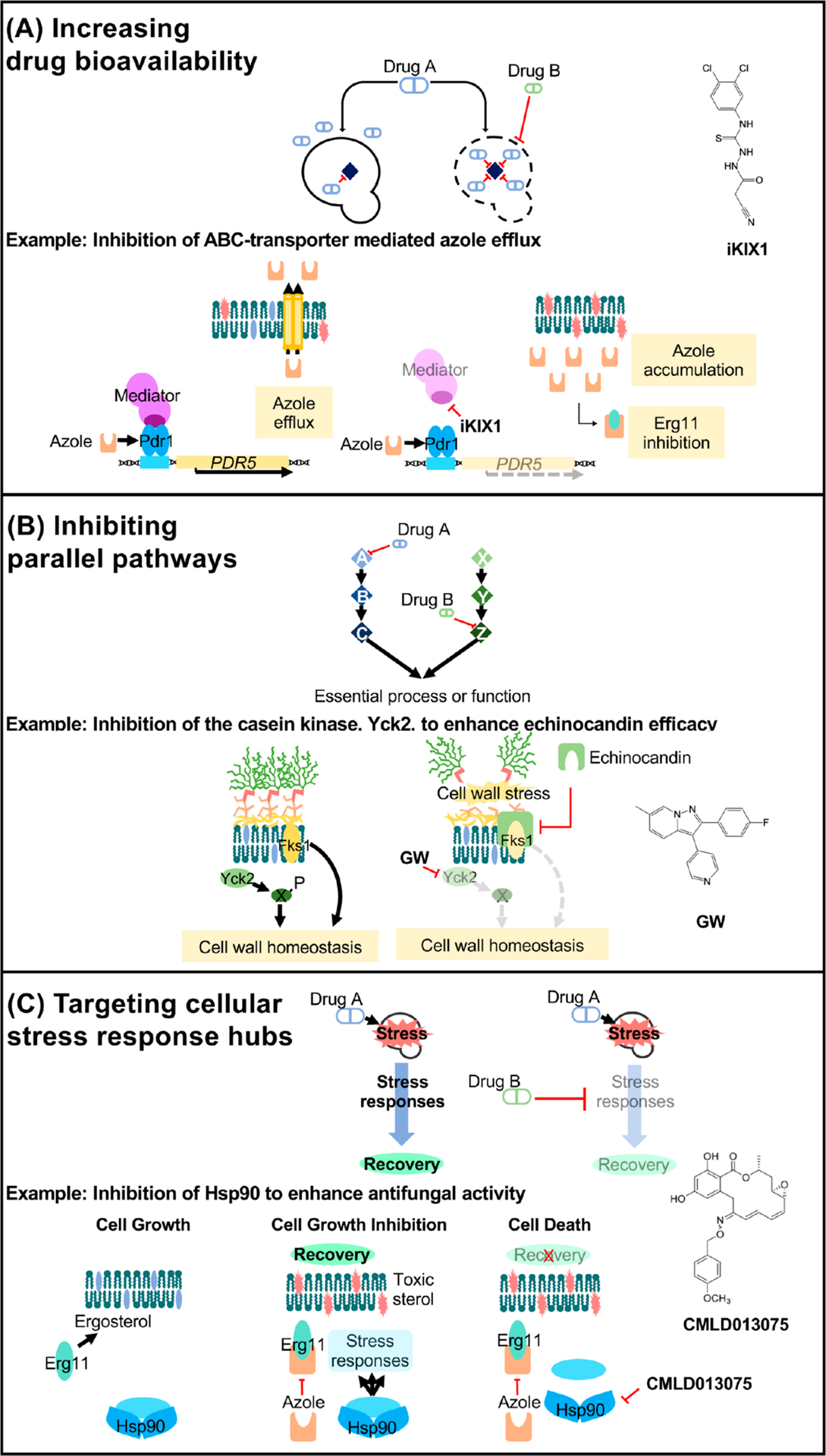
References
-
- Brown GD; Denning DW; Gow NA; Levitz SM; Netea MG; White TC Hidden Killers: Human Fungal Infections. Sci. Transl. Med 2012, 4, 165rv13. - PubMed
-
- Fisher MC; Hawkins NJ; Sanglard D; Gurr SJ Worldwide Emergence Of Resistance To Antifungal Drugs Challenges Human Health And Food Security. Science 2018, 360, 739–742. - PubMed
-
- Pfaller MA; Diekema DJ Epidemiology of Invasive Mycoses in North America. Crit. Rev. Microbiol 2010, 36, 1–53. - PubMed
-
- Enoch DA; Yang H; Aliyu SH; Micallef C The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol 2017, 1508, 17–65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
