

HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis
- PMID: 32442397
- PMCID: PMC7305998
- DOI: 10.1016/j.molcel.2020.04.031
HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis
Abstract
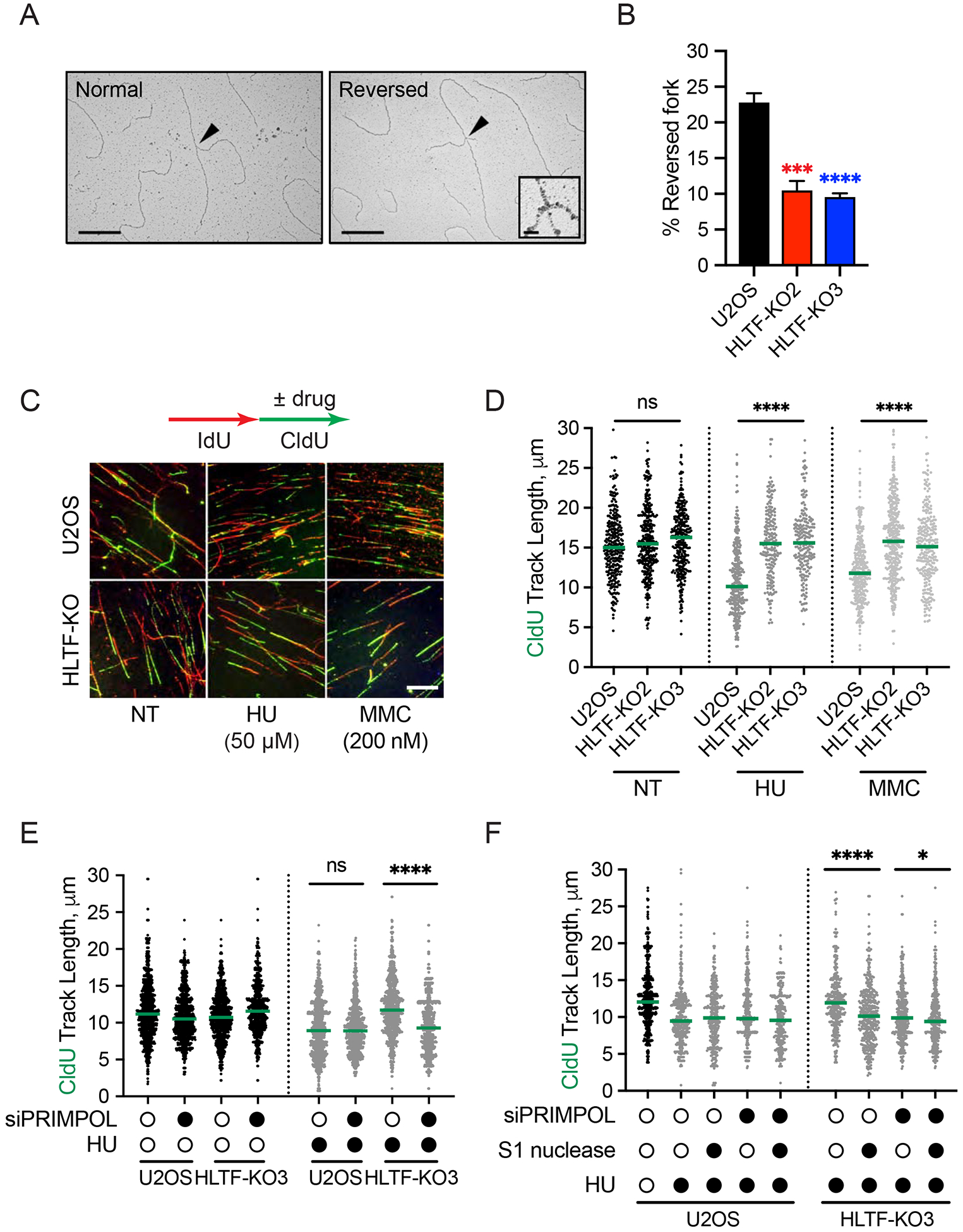
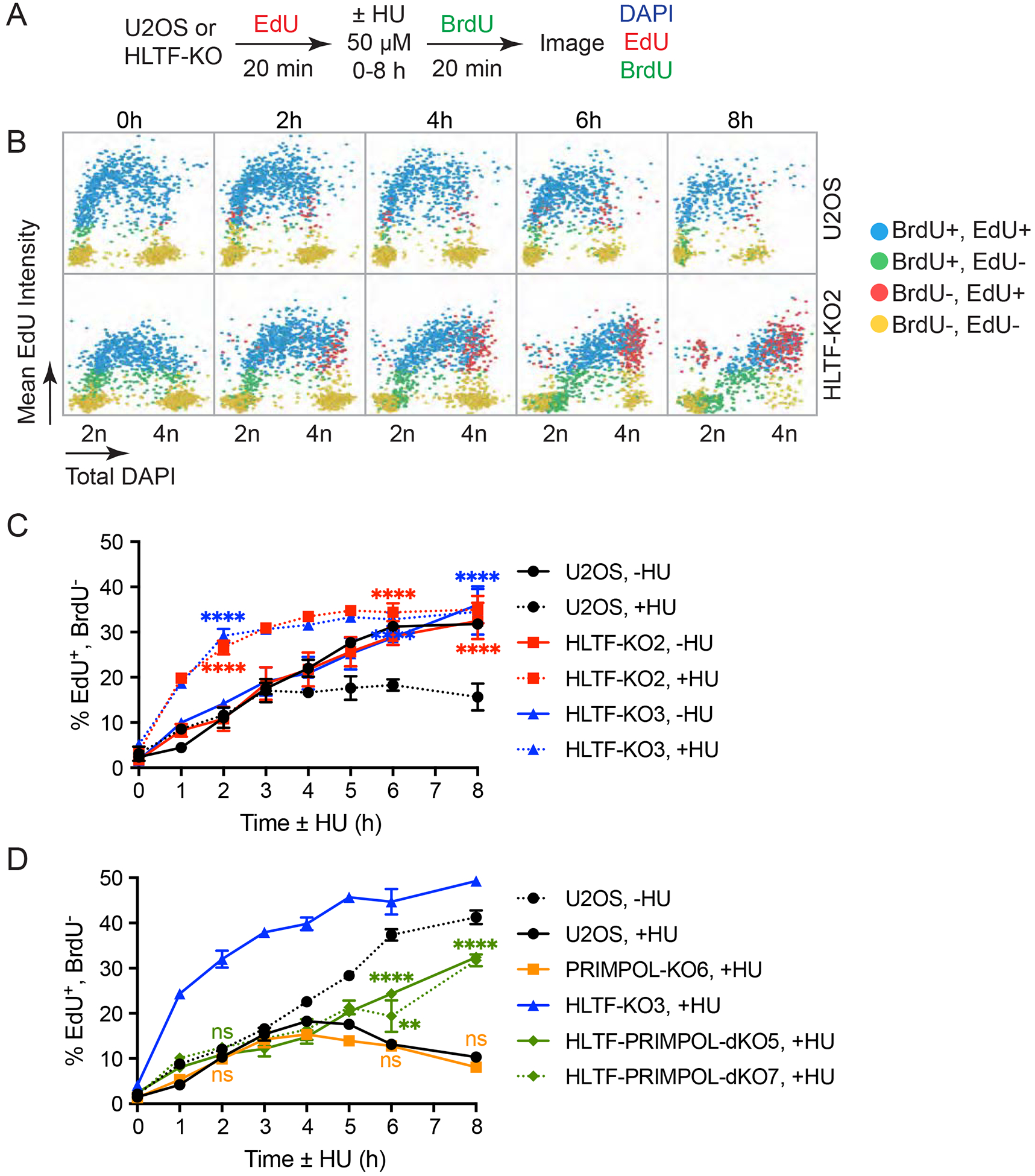
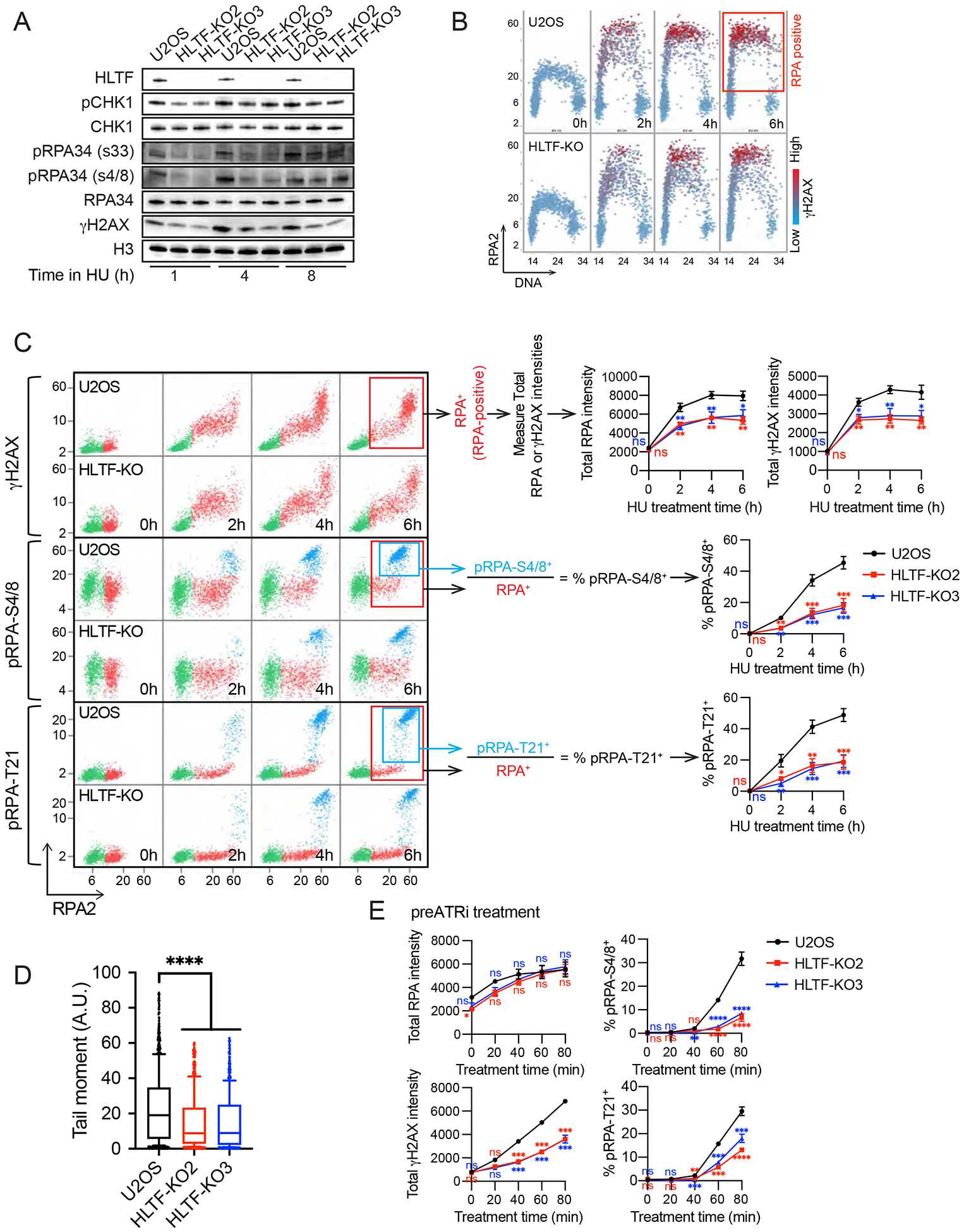
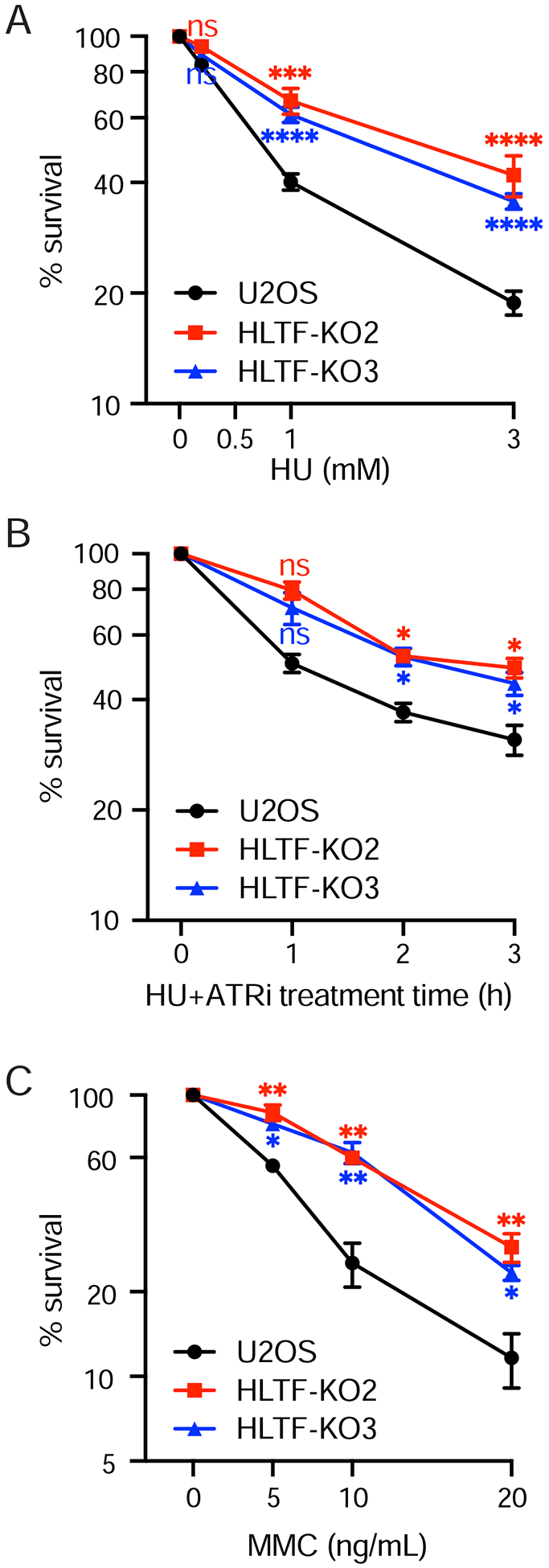
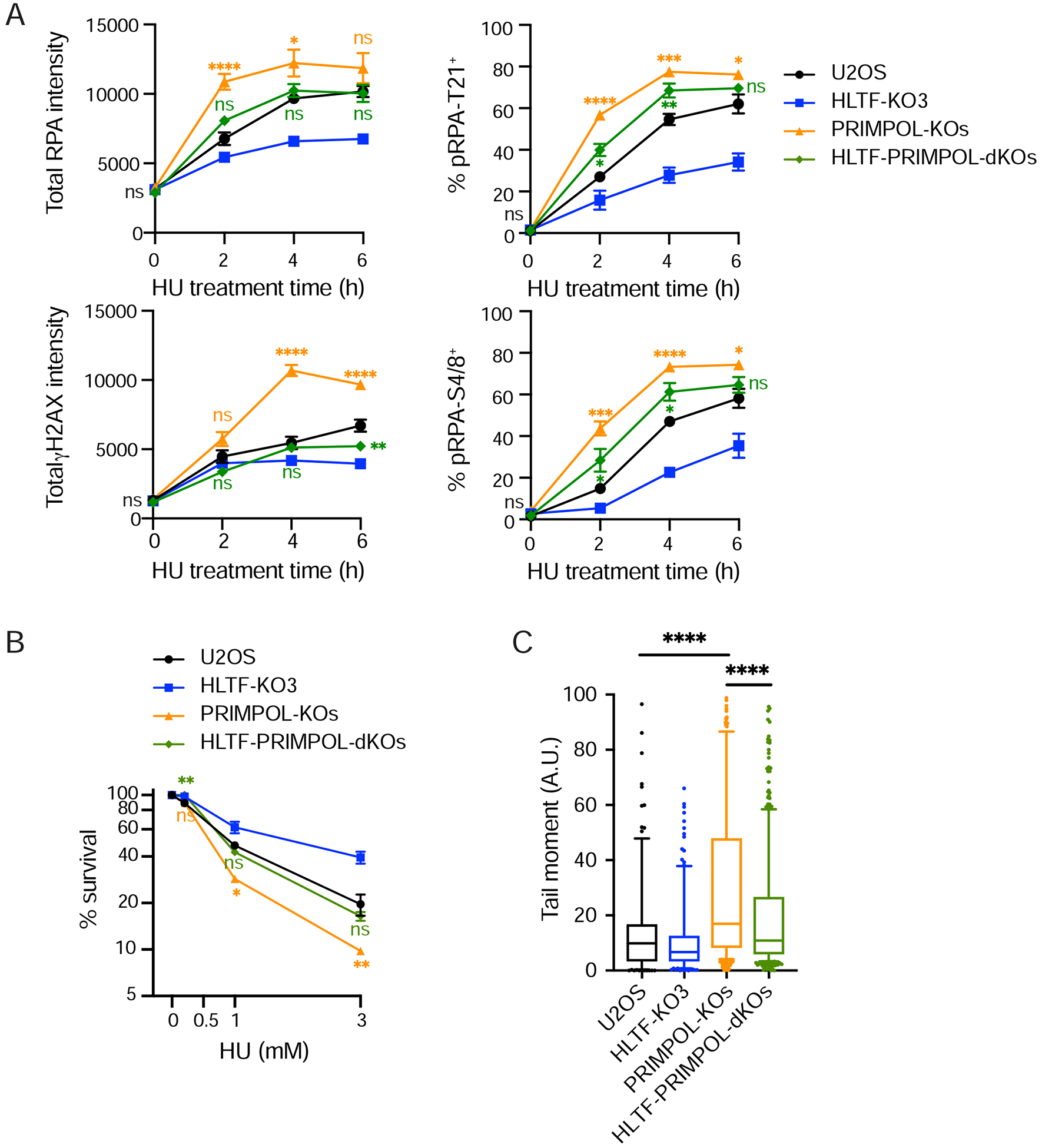
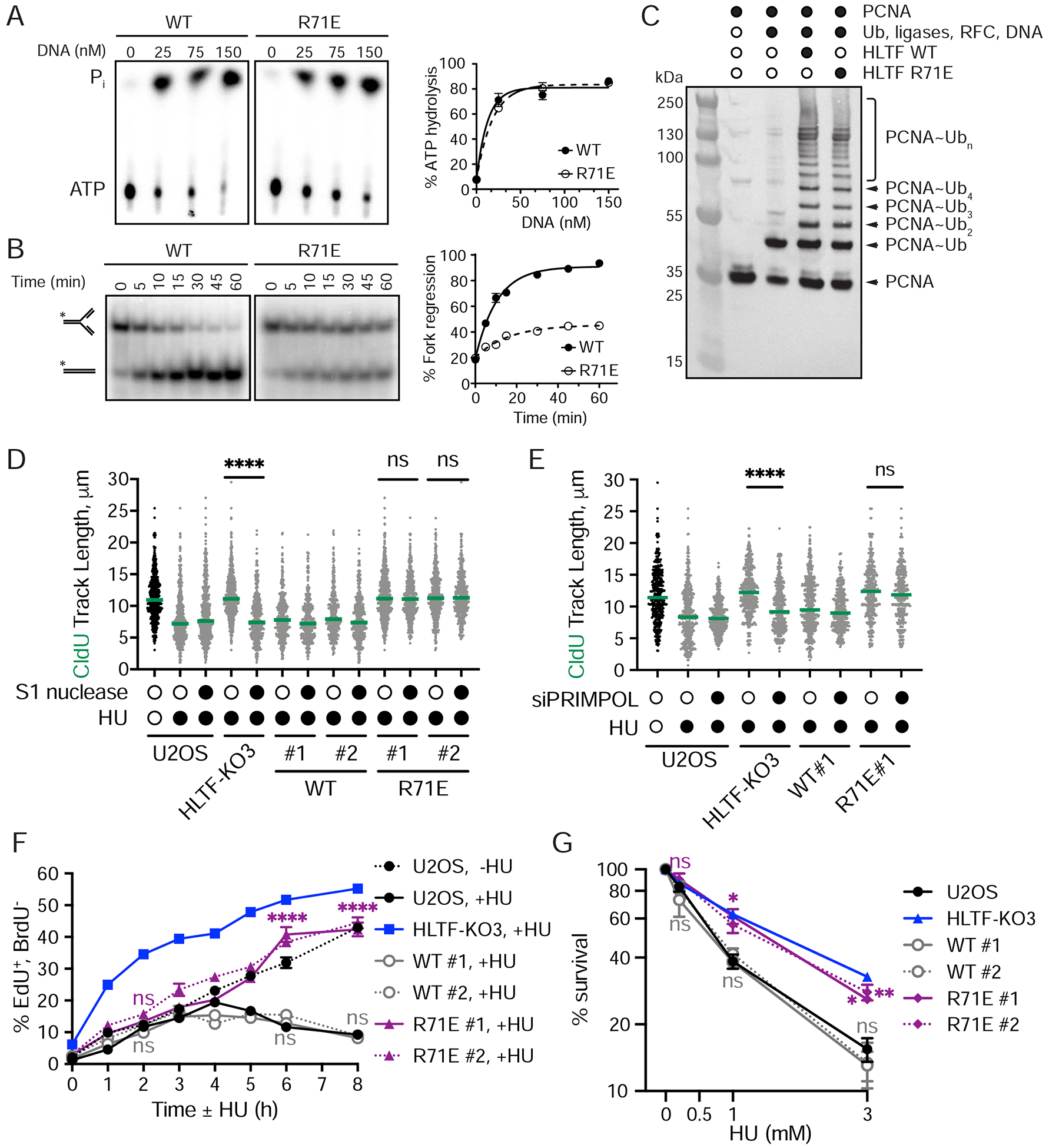
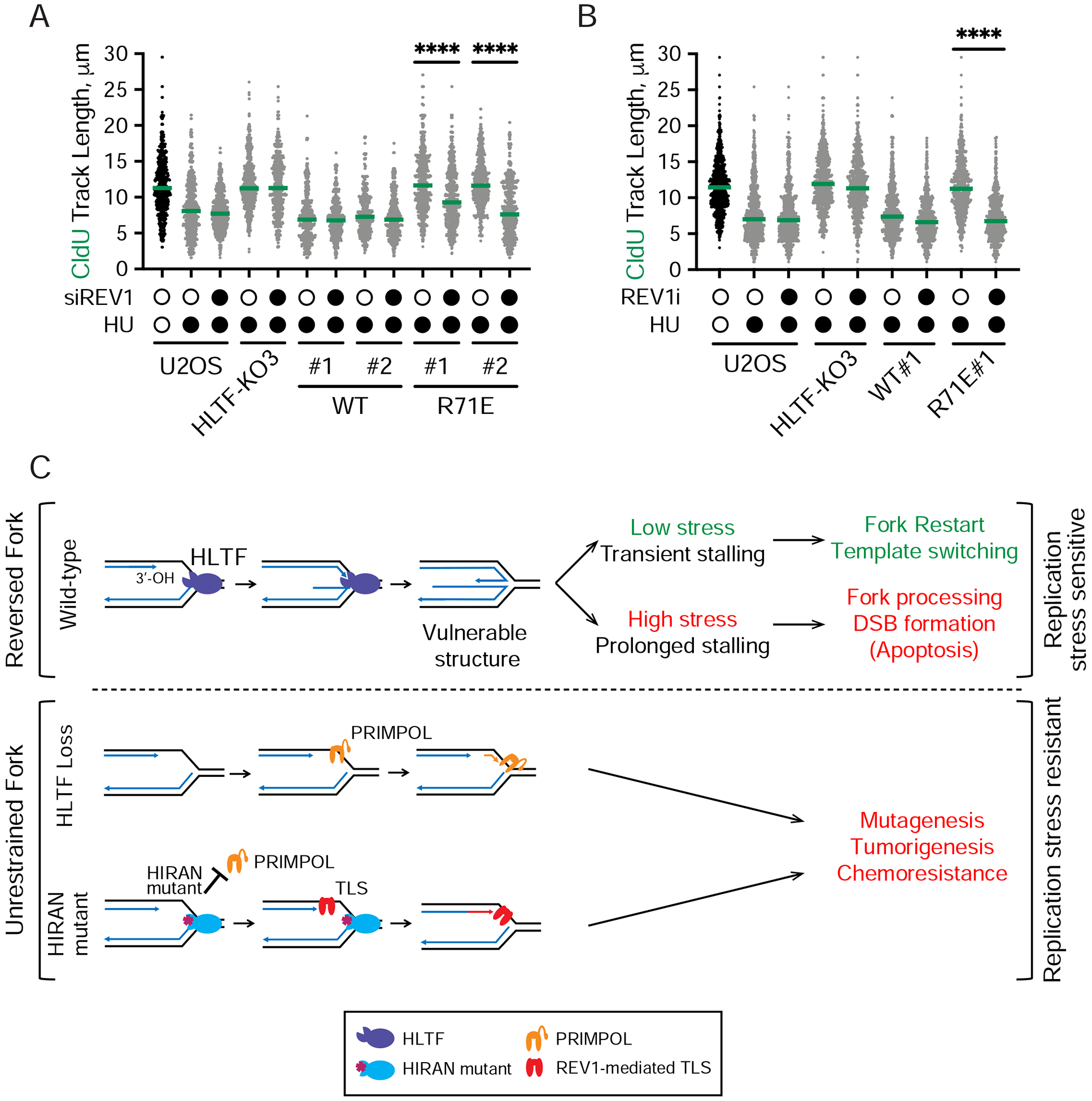
DNA replication stress can stall replication forks, leading to genome instability. DNA damage tolerance pathways assist fork progression, promoting replication fork reversal, translesion DNA synthesis (TLS), and repriming. In the absence of the fork remodeler HLTF, forks fail to slow following replication stress, but underlying mechanisms and cellular consequences remain elusive. Here, we demonstrate that HLTF-deficient cells fail to undergo fork reversal in vivo and rely on the primase-polymerase PRIMPOL for repriming, unrestrained replication, and S phase progression upon limiting nucleotide levels. By contrast, in an HLTF-HIRAN mutant, unrestrained replication relies on the TLS protein REV1. Importantly, HLTF-deficient cells also exhibit reduced double-strand break (DSB) formation and increased survival upon replication stress. Our findings suggest that HLTF promotes fork remodeling, preventing other mechanisms of replication stress tolerance in cancer cells. This remarkable plasticity of the replication fork may determine the outcome of replication stress in terms of genome integrity, tumorigenesis, and response to chemotherapy.
Keywords: DNA replication, replication stress response, fork reversal, HLTF, PRIMPOL, REV1, DNA damage tolerance, translesion synthesis, ATR inhibition, replication catastrophe.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
