

De Novo SOX6 Variants Cause a Neurodevelopmental Syndrome Associated with ADHD, Craniosynostosis, and Osteochondromas
- PMID: 32442410
- PMCID: PMC7273536
- DOI: 10.1016/j.ajhg.2020.04.015
De Novo SOX6 Variants Cause a Neurodevelopmental Syndrome Associated with ADHD, Craniosynostosis, and Osteochondromas
Abstract
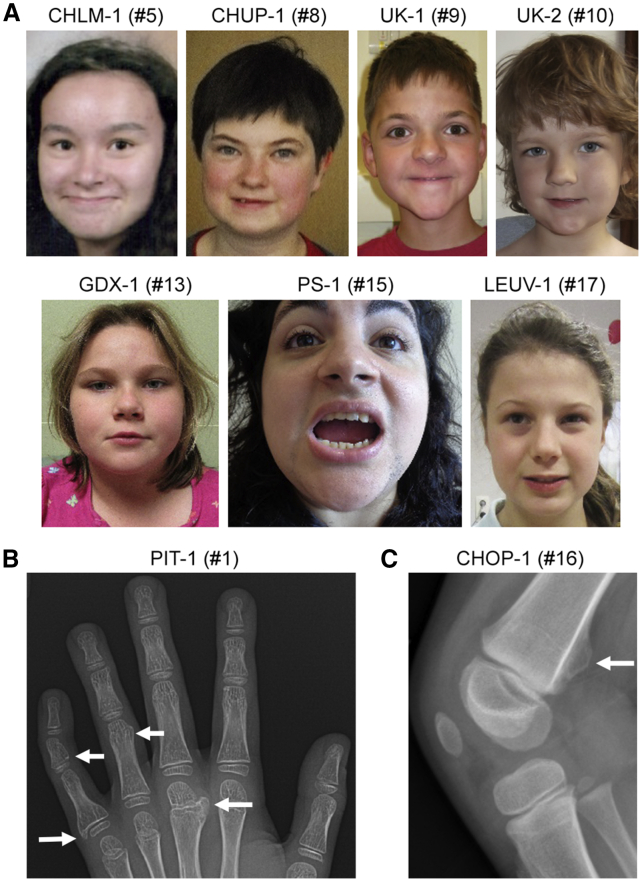
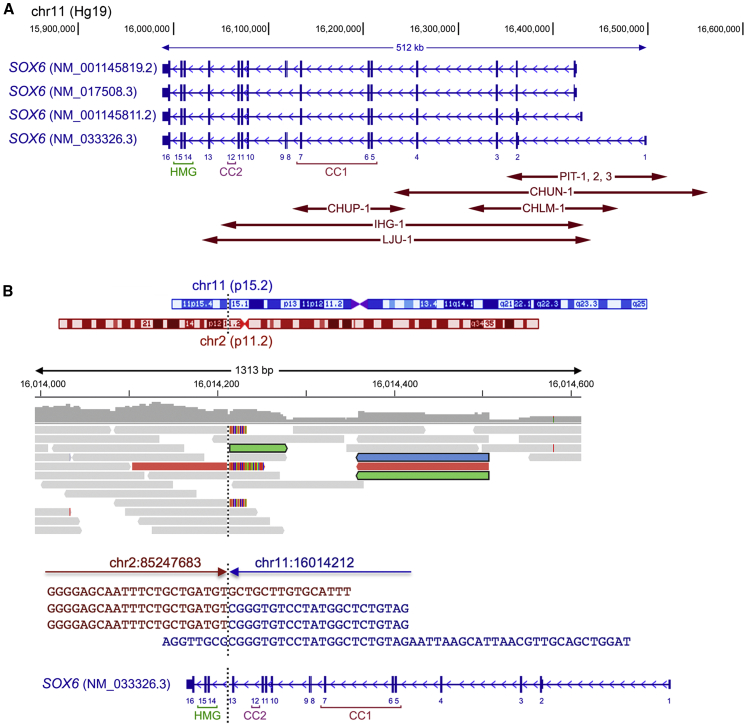
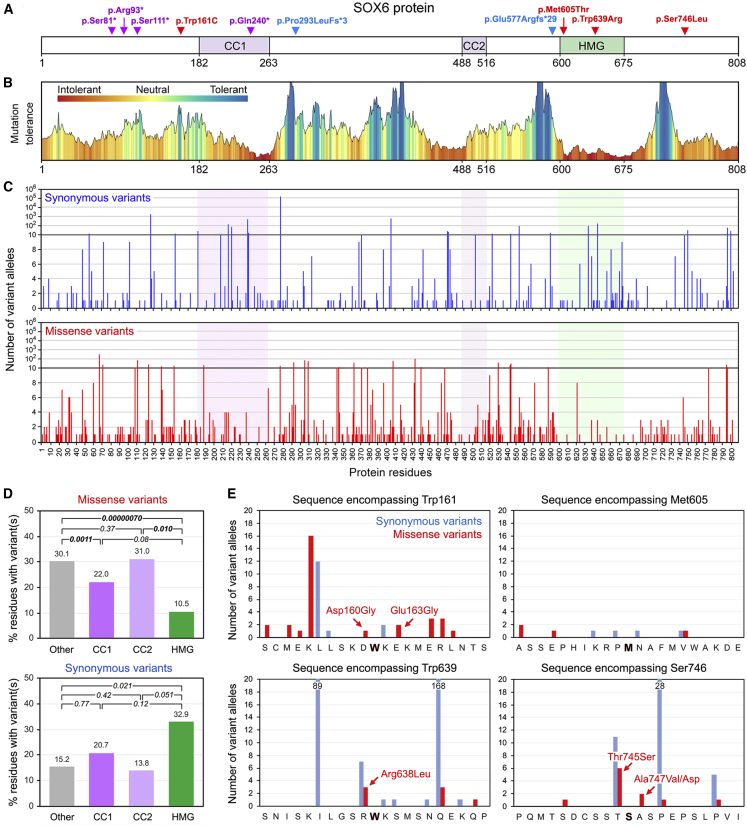
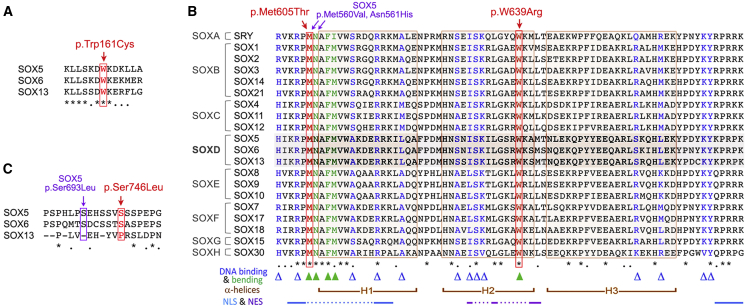
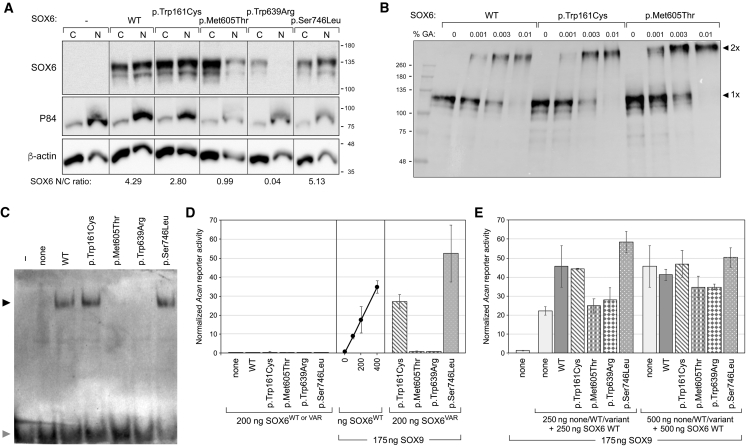
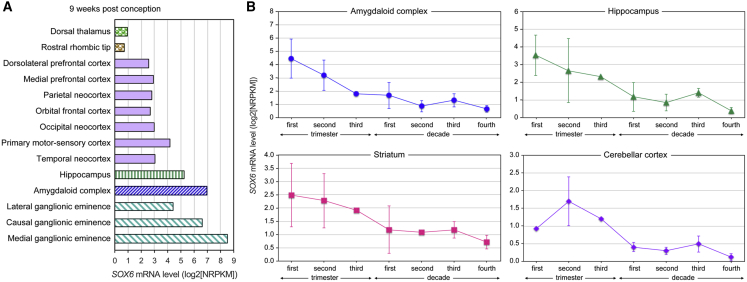
SOX6 belongs to a family of 20 SRY-related HMG-box-containing (SOX) genes that encode transcription factors controlling cell fate and differentiation in many developmental and adult processes. For SOX6, these processes include, but are not limited to, neurogenesis and skeletogenesis. Variants in half of the SOX genes have been shown to cause severe developmental and adult syndromes, referred to as SOXopathies. We here provide evidence that SOX6 variants also cause a SOXopathy. Using clinical and genetic data, we identify 19 individuals harboring various types of SOX6 alterations and exhibiting developmental delay and/or intellectual disability; the individuals are from 17 unrelated families. Additional, inconstant features include attention-deficit/hyperactivity disorder (ADHD), autism, mild facial dysmorphism, craniosynostosis, and multiple osteochondromas. All variants are heterozygous. Fourteen are de novo, one is inherited from a mosaic father, and four offspring from two families have a paternally inherited variant. Intragenic microdeletions, balanced structural rearrangements, frameshifts, and nonsense variants are predicted to inactivate the SOX6 variant allele. Four missense variants occur in residues and protein regions highly conserved evolutionarily. These variants are not detected in the gnomAD control cohort, and the amino acid substitutions are predicted to be damaging. Two of these variants are located in the HMG domain and abolish SOX6 transcriptional activity in vitro. No clear genotype-phenotype correlations are found. Taken together, these findings concur that SOX6 haploinsufficiency leads to a neurodevelopmental SOXopathy that often includes ADHD and abnormal skeletal and other features.
Keywords: ADHD; SOX6; SOXopathy; craniosynostosis; developmental delay; dysmorphism; genetic variant; human disease; intellectual disability; osteochondroma.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
A.B., C.F., L.B.H., and P.R. are employees of GeneDx. All other authors declare no competing interests.
Figures
References
-
- Kamachi Y., Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140:4129–4144. - PubMed
-
- Gubbay J., Collignon J., Koopman P., Capel B., Economou A., Münsterberg A., Vivian N., Goodfellow P., Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. - PubMed
-
- Sinclair A.H., Berta P., Palmer M.S., Hawkins J.R., Griffiths B.L., Smith M.J., Foster J.W., Frischauf A.M., Lovell-Badge R., Goodfellow P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240–244. - PubMed
-
- Kashimada K., Koopman P. Sry: the master switch in mammalian sex determination. Development. 2010;137:3921–3930. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
