

Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways
- PMID: 32443586
- PMCID: PMC7291004
- DOI: 10.3390/genes11050565
Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways
Abstract
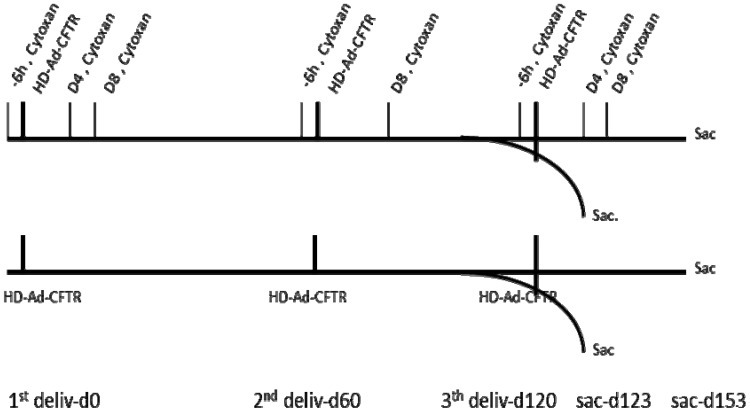
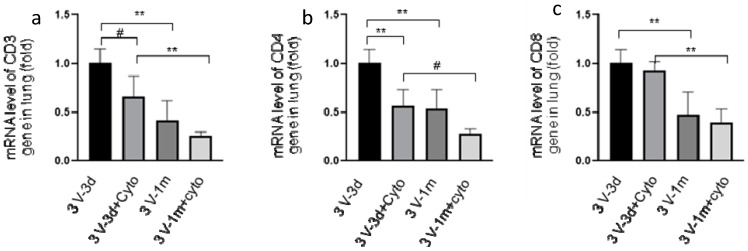
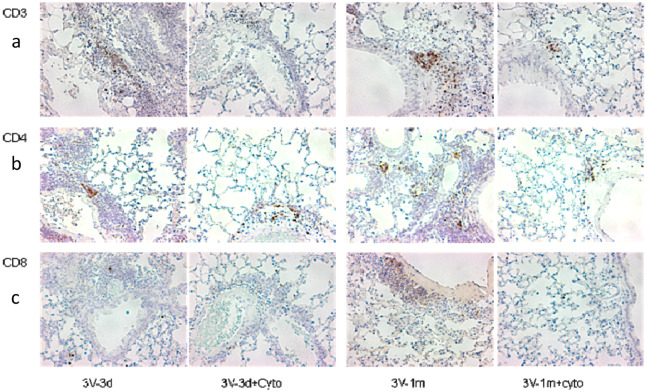
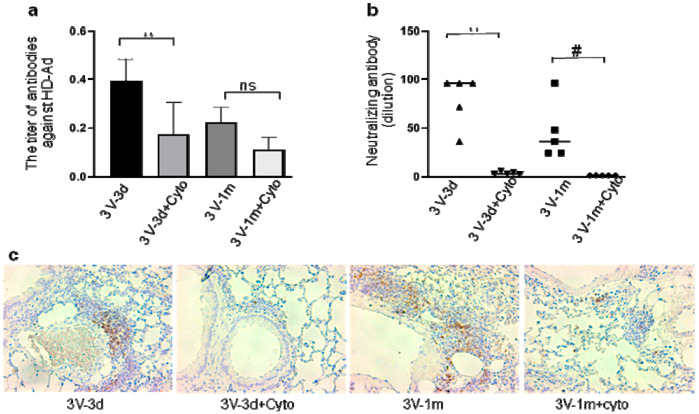
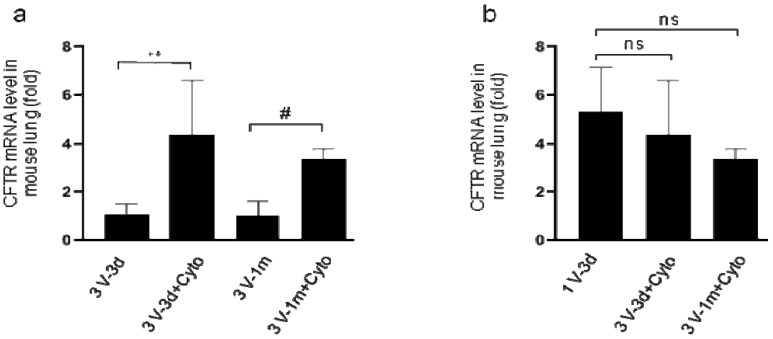
Cystic Fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, and CF patients require life-long treatment. Although CFTR modulators show a great potential for treating most CF patients, some individuals may not tolerate the treatment. In addition, there is no effective therapy for patients with some rare CFTR mutations, such as class I CF mutations, which lead to a lack of CFTR protein production. Therefore, other therapeutic strategies, such as gene therapy, have to be investigated. Currently, immune responses to gene therapy vectors and transgene products are a major obstacle to applying CF gene therapy to clinical applications. In this study, we examined the effects of cyclophosphamide on the modulation of host immune responses and for the improvement of the CFTR transgene expression in the repeated delivery of helper-dependent adenoviral (HD-Ad) vectors to mouse lungs. We have found that cyclophosphamide significantly decreased the expression of T cell genes, such as CD3 (cluster of differentiation 3) and CD4, and reduced their infiltration into mouse lung tissues. We have also found that the levels of the anti-adenoviral antibody and neutralizing activity as well as B-cell infiltration into the mouse lung tissues were significantly reduced with this treatment. Correspondingly, the expression of the human CFTR transgene has been significantly improved with cyclophosphamide administration compared to the group with no treatment. These data suggest that the sustained expression of the human CFTR transgene in mouse lungs through repeated vector delivery can be achieved by transient immunosuppression.
Keywords: cyclophosphamide; cystic fibrosis; gene therapy; transient immunosuppression.
Conflict of interest statement
All authors have no conflict of interest to declare.
Figures
References
-
- Davies J.C., Moskowitz S.M., Brown C., Horsley A., Mall M.A., McKone E.F., Plant B.J., Prais D., Ramsey B.W., Taylor-Cousar J.L., et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018;379:1599–1611. doi: 10.1056/NEJMoa1807119. - DOI - PMC - PubMed
-
- Heijerman H.G.M., McKone E.F., Downey D.G., Van Braeckel E., Rowe S.M., Tullis E., Mall M.A., Welter J.J., Ramsey B.W., McKee C.M., et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. - DOI - PMC - PubMed
-
- Keating D., Marigowda G., Burr L., Daines C., Mall M.A., McKone E.F., Ramsey B.W., Rowe S.M., Sass L.A., Tullis E., et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018;379:1612–1620. doi: 10.1056/NEJMoa1807120. - DOI - PMC - PubMed
-
- Middleton P.G., Mall M.A., Drevinek P., Lands L.C., McKone E.F., Polineni D., Ramsey B.W., Taylor-Cousar J.L., Tullis E., Vermeulen F., et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
