

Enhancer Predictions and Genome-Wide Regulatory Circuits
- PMID: 32443951
- PMCID: PMC7644210
- DOI: 10.1146/annurev-genom-121719-010946
Enhancer Predictions and Genome-Wide Regulatory Circuits
Abstract
Spatiotemporal control of gene expression during development requires orchestrated activities of numerous enhancers, which are cis-regulatory DNA sequences that, when bound by transcription factors, support selective activation or repression of associated genes. Proper activation of enhancers is critical during embryonic development, adult tissue homeostasis, and regeneration, and inappropriate enhancer activity is often associated with pathological conditions such as cancer. Multiple consortia [e.g., the Encyclopedia of DNA Elements (ENCODE) Consortium and National Institutes of Health Roadmap Epigenomics Mapping Consortium] and independent investigators have mapped putative regulatory regions in a large number of cell types and tissues, but the sequence determinants of cell-specific enhancers are not yet fully understood. Machine learning approaches trained on large sets of these regulatory regions can identify core transcription factor binding sites and generate quantitative predictions of enhancer activity and the impact of sequence variants on activity. Here, we review these computational methods in the context of enhancer prediction and gene regulatory network models specifying cell fate.
Keywords: cell fate switching; enhancers; gene regulatory networks; machine learning; sequence-based prediction.
Figures
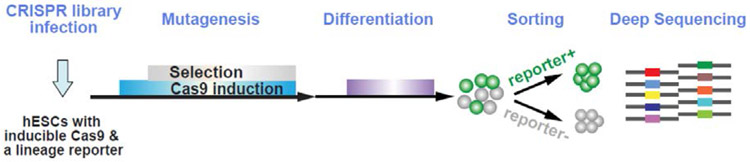




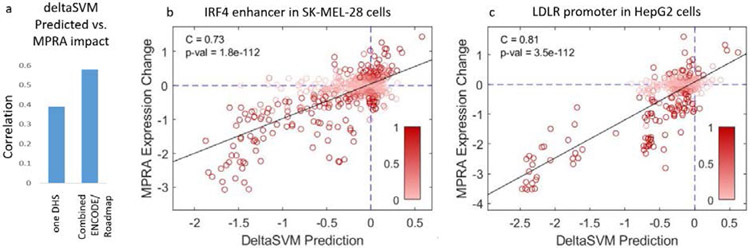
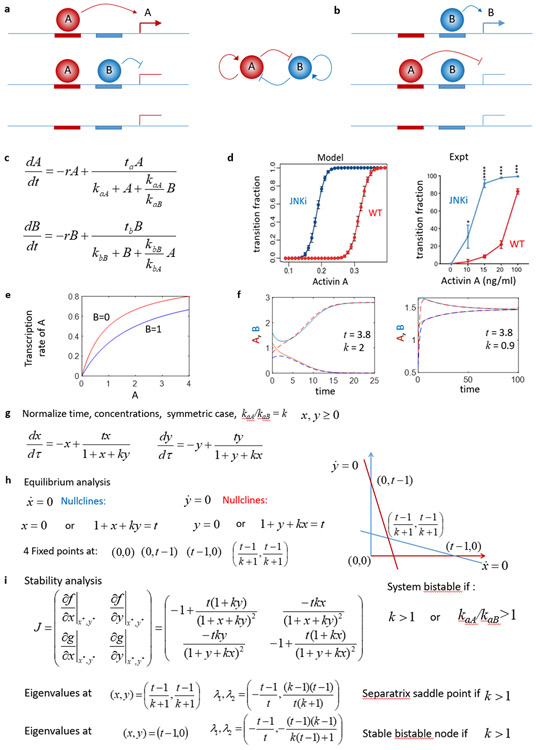
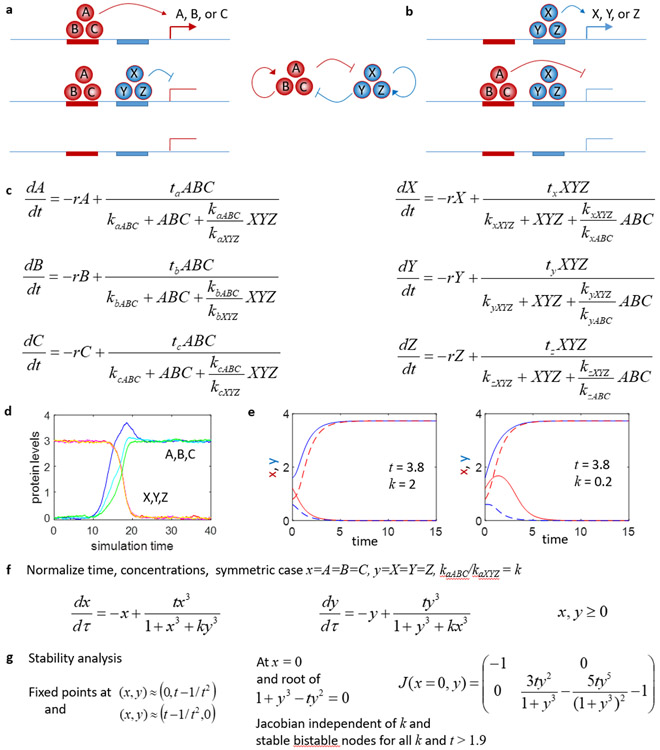
References
-
- Alexander J, Stainier DYR. 1999. A molecular pathway leading to endoderm formation in zebrafish. Current Biology. 9(20):1147–57 - PubMed
-
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. 2015. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotech. 33(8):831–38 - PubMed
-
- Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nature Reviews Genetics. 17(8):487–500 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
