

Ancient founder mutation in RUBCN: a second unrelated family confirms Salih ataxia (SCAR15)
- PMID: 32450808
- PMCID: PMC7249383
- DOI: 10.1186/s12883-020-01761-w
Ancient founder mutation in RUBCN: a second unrelated family confirms Salih ataxia (SCAR15)
Abstract
Background: Homozygous frameshift mutation in RUBCN (KIAA0226), known to result in endolysosomal machinery defects, has previously been reported in a single Saudi family with autosomal recessive spinocerebellar ataxia (Salih ataxia, SCAR15, OMIM # 615705). The present report describes the clinical, neurophysiologic, neuroimaging, and genetic findings in a second unrelated Saudi family with two affected children harboring identical homozygous frameshift mutation in the gene. It also explores and documents an ancient founder cerebellar ataxia mutation in the Arabian Peninsula.
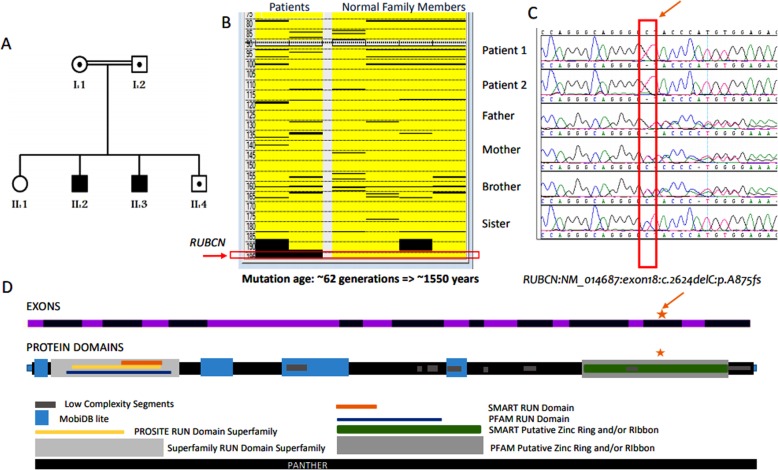
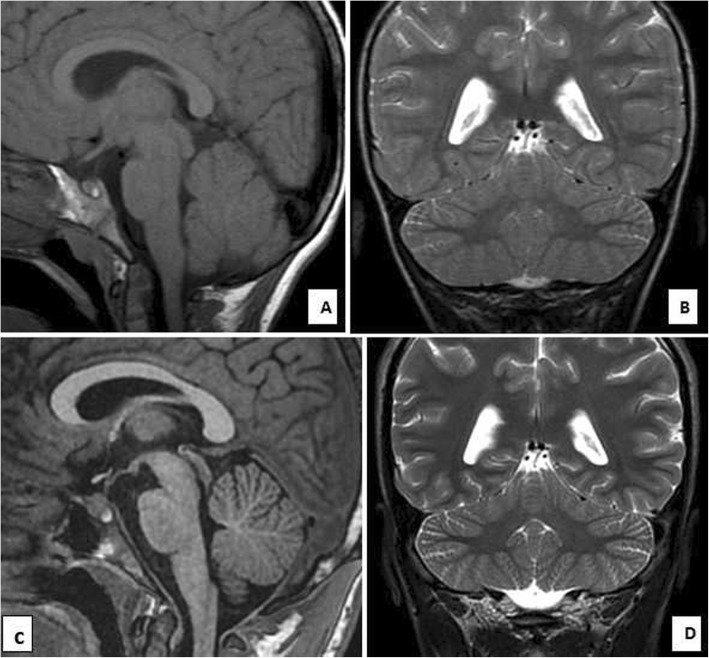
Case presentation: The present family has two affected males (aged 6.5 and 17 years) with unsteady gait apparent since learning to walk at 2.5 and 3 years, respectively. The younger patient showed gait ataxia and normal reflexes. The older patient had saccadic eye movement, dysarthria, mild upper and lower limb and gait ataxia (on tandem walking), and enhanced reflexes in the lower limbs. Cognitive abilities were mildly impaired in the younger sibling (IQ 67) and borderline in the older patient (IQ 72). Nerve conduction studies were normal in both patients. MRI was normal at 2.5 years in the younger sibling. Brain MRI showed normal cerebellar volume and folia in the older sibling at the age of 6 years, and revealed minimal superior vermian atrophy at the age of 16 years. Autozygome and exome analysis showed both affected have previously reported homoallelic mutation in RUBCN (NM_014687:exon18:c.2624delC:p.A875fs), whereas the parents are carriers. Autozygosity mapping focused on smallest haplotype on chromosome 3 and mutation age analysis revealed the mutation occurred approximately 1550 years ago spanning about 62 generations.
Conclusions: Our findings validate the slowly progressive phenotype of Salih ataxia (SCAR15, OMIM # 615705) by an additional family. Haplotype sharing attests to a common founder, an ancient RUBCN mutation in the Arab population.
Keywords: Founder mutation; RUBCN; SCAR15; Salih ataxia.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Assoum M, Salih MA, Drouot N, H'Mida-Ben Brahim D, Lagier-Tourenne C, AlDrees A, Elmalik SA, Ahmed TS, Seidahmed MZ, Kabiraj MM, et al. Rundataxin, a novel protein with RUN and diacylglycerol binding domains, is mutant in a new recessive ataxia. Brain. 2010;133(Pt 8):2439–2447. doi: 10.1093/brain/awq181. - DOI - PubMed
-
- Al-Sayed MD, Al-Zaidan H, Albakheet A, Hakami H, Kenana R, Al-Yafee Y, Al-Dosary M, Qari A, Al-Sheddi T, Al-Muheiza M, et al. Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet. 2013;93(4):721–726. doi: 10.1016/j.ajhg.2013.08.001. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
