

An Update on XMEN Disease
- PMID: 32451662
- PMCID: PMC7369250
- DOI: 10.1007/s10875-020-00790-x
An Update on XMEN Disease
Abstract
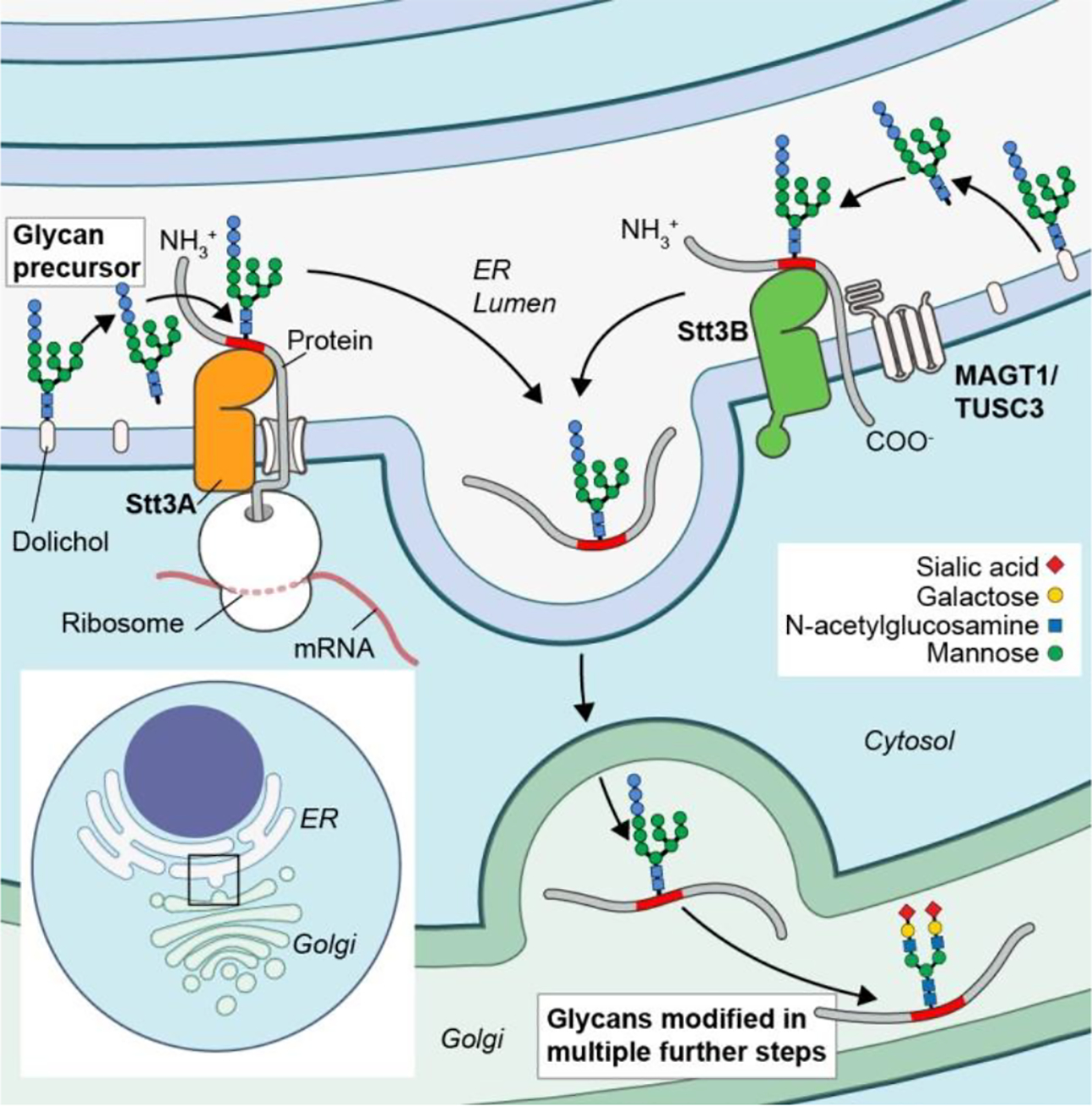
"X-linked immunodeficiency with magnesium defect, Epstein-Barr virus (EBV) infection, and neoplasia" (XMEN) disease is an inborn error of glycosylation and immunity caused by loss of function mutations in the magnesium transporter 1 (MAGT1) gene. It is a multisystem disease that strongly affects certain immune cells. MAGT1 is now confirmed as a non-catalytic subunit of the oligosaccharyltransferase complex and facilitates Asparagine (N)-linked glycosylation of specific substrates, making XMEN a congenital disorder of glycosylation manifesting as a combined immune deficiency. The clinical disease has variable expressivity, and impaired glycosylation of key MAGT1-dependent glycoproteins in addition to Mg2+ abnormalities can explain some of the immune manifestations. NKG2D, an activating receptor critical for cytotoxic function against EBV, is poorly glycosylated and invariably decreased on CD8+ T cells and natural killer (NK) cells from XMEN patients. It is the best biomarker of the disease. The characterization of EBV-naïve XMEN patients has clarified features of the genetic disease that were previously attributed to EBV infection. Extra-immune manifestations, including hepatic and neurological abnormalities, have recently been reported. EBV-associated lymphomas remain the main cause of severe morbidity. Unfortunately, treatment options to address the underlying mechanism of disease remain limited and Mg2+ supplementation has not proven successful. Here, we review the expanding clinical phenotype and recent advances in glycobiology that have increased our understanding of XMEN disease. We also propose updating XMEN to "X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect" in light of these novel findings.
Keywords: CD28; CD70; Epstein-Barr virus; Immunodeficiency; MAGT1; NKG2D; XMEN disease; carbohydrate deficient transferrin; congenital disorders of glycosylation; lymphoma; magnesium; oligosaccharyltransferase complex.
Conflict of interest statement
The authors declared that they have no conflict of interest
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
