

Congenital hearing impairment associated with peripheral cochlear nerve dysmyelination in glycosylation-deficient muscular dystrophy
- PMID: 32453729
- PMCID: PMC7274486
- DOI: 10.1371/journal.pgen.1008826
Congenital hearing impairment associated with peripheral cochlear nerve dysmyelination in glycosylation-deficient muscular dystrophy
Abstract
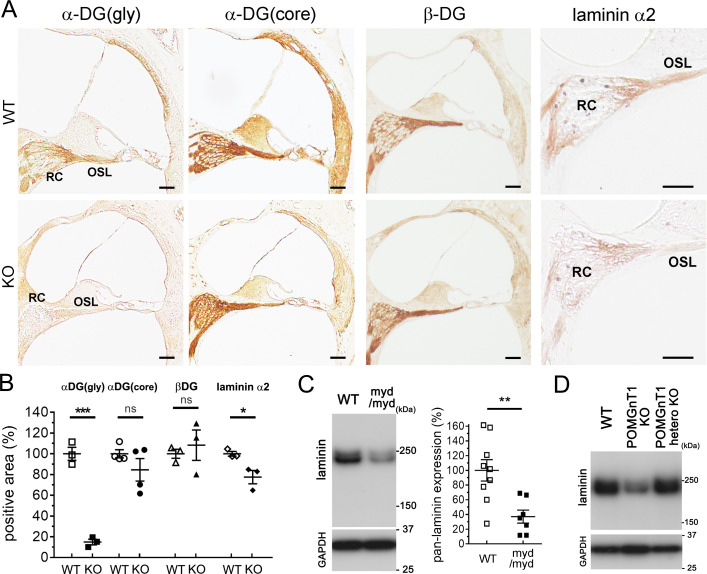
Hearing loss (HL) is one of the most common sensory impairments and etiologically and genetically heterogeneous disorders in humans. Muscular dystrophies (MDs) are neuromuscular disorders characterized by progressive degeneration of skeletal muscle accompanied by non-muscular symptoms. Aberrant glycosylation of α-dystroglycan causes at least eighteen subtypes of MD, now categorized as MD-dystroglycanopathy (MD-DG), with a wide spectrum of non-muscular symptoms. Despite a growing number of MD-DG subtypes and increasing evidence regarding their molecular pathogeneses, no comprehensive study has investigated sensorineural HL (SNHL) in MD-DG. Here, we found that two mouse models of MD-DG, Largemyd/myd and POMGnT1-KO mice, exhibited congenital, non-progressive, and mild-to-moderate SNHL in auditory brainstem response (ABR) accompanied by extended latency of wave I. Profoundly abnormal myelination was found at the peripheral segment of the cochlear nerve, which is rich in the glycosylated α-dystroglycan-laminin complex and demarcated by "the glial dome." In addition, patients with Fukuyama congenital MD, a type of MD-DG, also had latent SNHL with extended latency of wave I in ABR. Collectively, these findings indicate that hearing impairment associated with impaired Schwann cell-mediated myelination at the peripheral segment of the cochlear nerve is a notable symptom of MD-DG.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures







References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
