

Molecular, Evolutionary, and Structural Analysis of the Terminal Protein Domain of Hepatitis B Virus Polymerase, a Potential Drug Target
- PMID: 32455999
- PMCID: PMC7291194
- DOI: 10.3390/v12050570
Molecular, Evolutionary, and Structural Analysis of the Terminal Protein Domain of Hepatitis B Virus Polymerase, a Potential Drug Target
Abstract
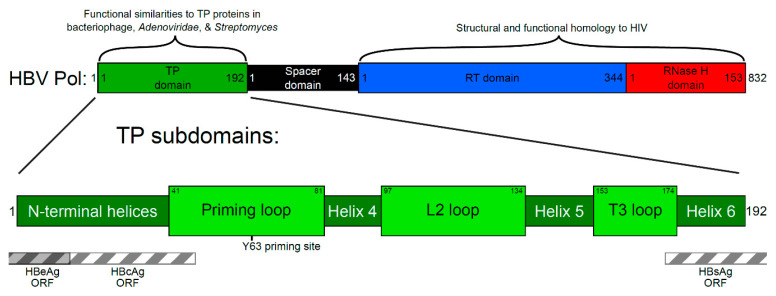
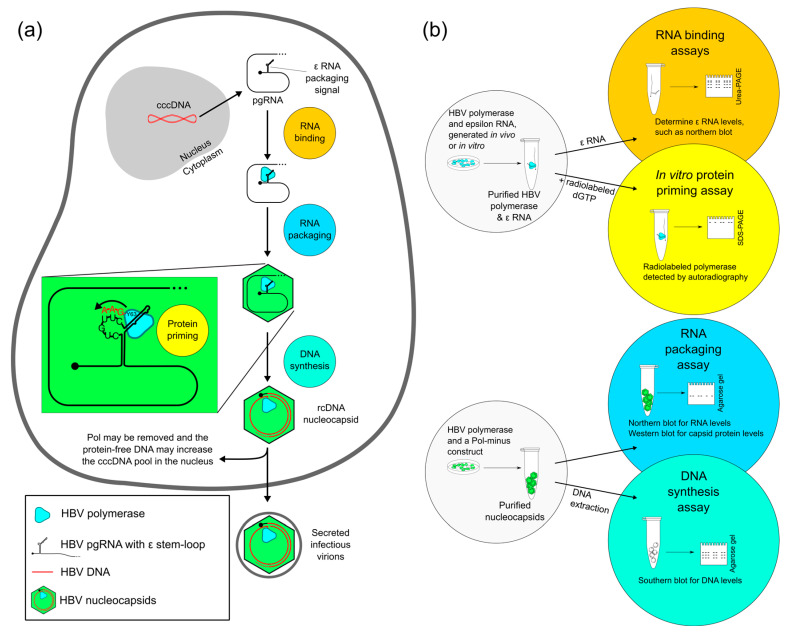
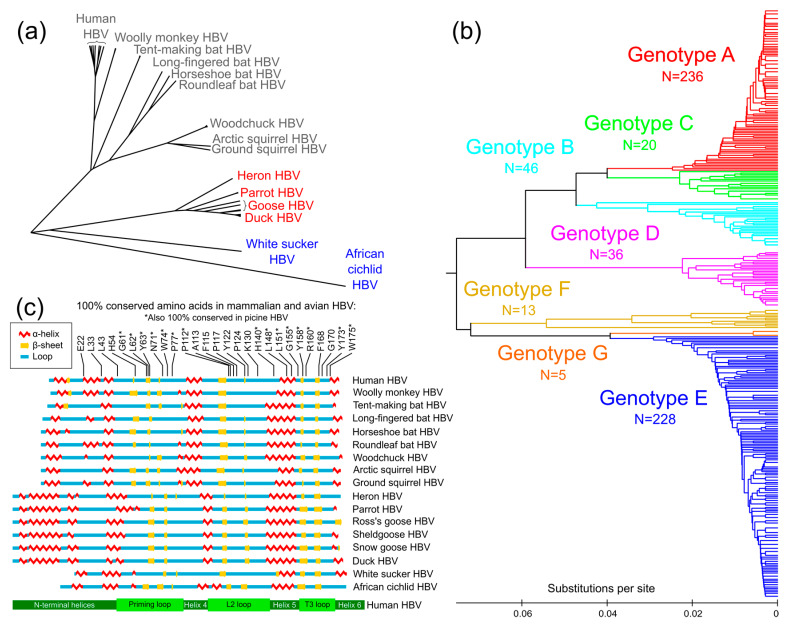
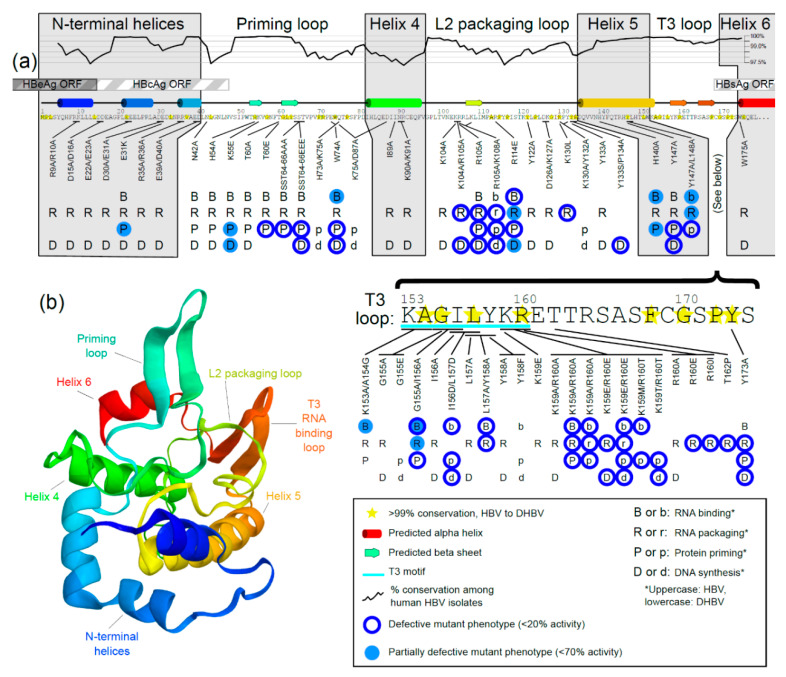
Approximately 250 million people are living with chronic hepatitis B virus (HBV) infections, which claim nearly a million lives annually. The target of all current HBV drug therapies (except interferon) is the viral polymerase; specifically, the reverse transcriptase domain. Although no high-resolution structure exists for the HBV polymerase, several recent advances have helped to map its functions to specific domains. The terminal protein (TP) domain, unique to hepadnaviruses such as HBV, has been implicated in the binding and packaging of the viral RNA, as well as the initial priming of and downstream synthesis of viral DNA-all of which make the TP domain an attractive novel drug target. This review encompasses three types of analysis: sequence conservation analysis, secondary structure prediction, and the results from mutational studies. It is concluded that the TP domain of HBV polymerase is comprised of seven subdomains (three unstructured loops and four helical regions) and that all three loop subdomains and Helix 5 are the major determinants of HBV function within the TP domain. Further studies, such as modeling inhibitors of these critical TP subdomains, will advance the TP domain of HBV polymerase as a therapeutic drug target in the progression towards a cure.
Keywords: hepatitis B virus; protein priming; terminal protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Smith S., Harmanci H., Hutin Y., Hess S., Bulterys M., Peck R., Rewari B., Mozalevskis A., Shibeshi M., Mumba M. Global progress on the elimination of viral hepatitis as a major public health threat: An analysis of WHO member state responses 2017. JHEP Rep. 2019;1:81–89. doi: 10.1016/j.jhepr.2019.04.002. - DOI - PMC - PubMed
-
- Hepatitis B Foundation: Drug Watch. [(accessed on 21 May 2020)]; Available online: http://www.hepb.org/treatment-and-management/drug-watch/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
