

Gene Panel Analysis in a Large Cohort of Patients With Autosomal Dominant Polycystic Kidney Disease Allows the Identification of 80 Potentially Causative Novel Variants and the Characterization of a Complex Genetic Architecture in a Subset of Families
- PMID: 32457805
- PMCID: PMC7224062
- DOI: 10.3389/fgene.2020.00464
Gene Panel Analysis in a Large Cohort of Patients With Autosomal Dominant Polycystic Kidney Disease Allows the Identification of 80 Potentially Causative Novel Variants and the Characterization of a Complex Genetic Architecture in a Subset of Families
Abstract
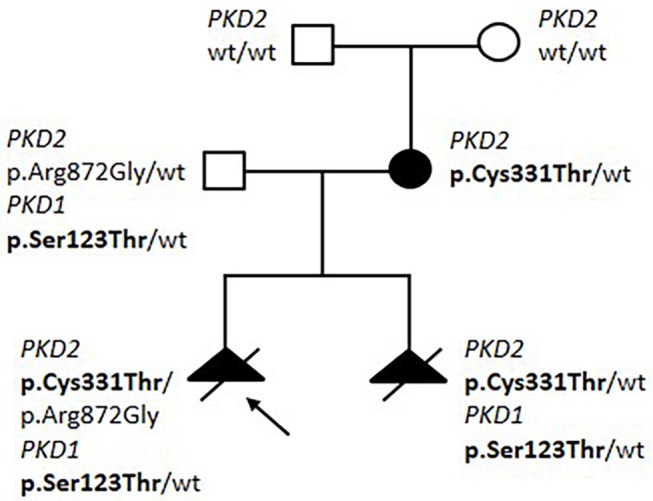
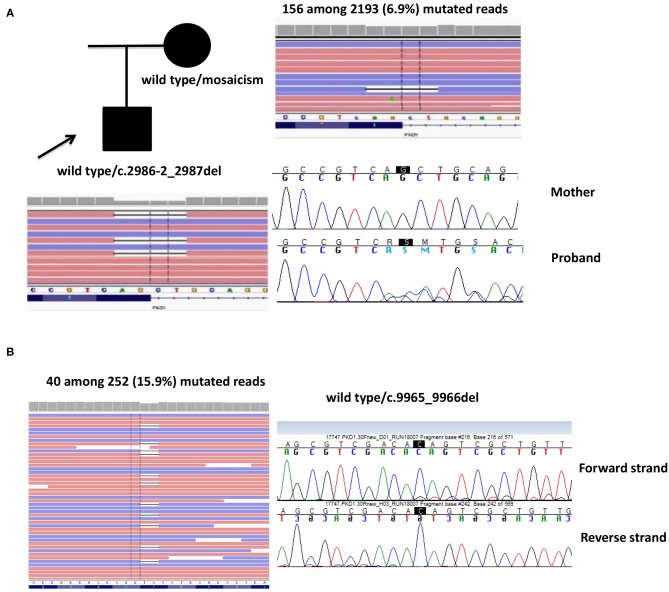
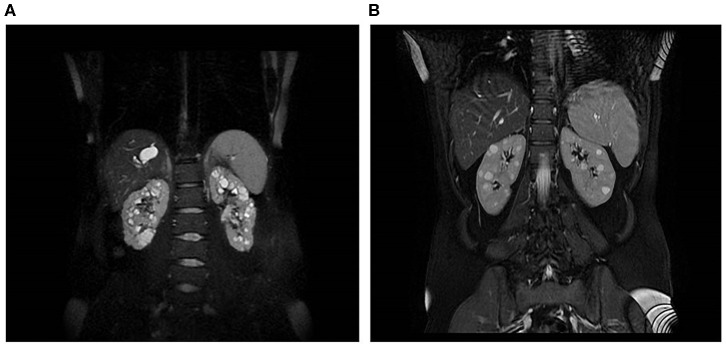
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited disorders in humans and the majority of patients carry a variant in either PKD1 or PKD2. Genetic testing is increasingly required for diagnosis, prognosis, and treatment decision, but it is challenging due to segmental duplications of PKD1, genetic and allelic heterogeneity, and the presence of many variants hypomorphic or of uncertain significance. We propose an NGS-based testing strategy for molecular analysis of ADPKD and its phenocopies, validated in a diagnostic setting. Materials and Methods: Our protocol is based on high-throughput simultaneous sequencing of PKD1 and PKD2 after long range PCR of coding regions, followed by a masked reference genome alignment, and MLPA analysis. A further screening of additional 14 cystogenes was performed in negative cases. We applied this strategy to analyze 212 patients with a clinical suspicion of ADPKD. Results and Discussion: We detected causative variants (interpreted as pathogenic/likely pathogenic) in 61.3% of our index patients, and variants of uncertain clinical significance in 12.5%. The majority (88%) of genetic variants was identified in PKD1, 12% in PKD2. Among 158 distinct variants, 80 (50.6%) were previously unreported, confirming broad allelic heterogeneity. Eleven patients showed more than one variant. Segregation analysis indicated biallelic disease in five patients, digenic in one, de novo variant with unknown phase in two. Furthermore, our NGS protocol allowed the identification of two patients with somatic mosaicism, which was undetectable with Sanger sequencing. Among patients without PKD1/PKD2 variants, we identified three with possible alternative diagnosis: a patient with biallelic mutations in PKHD1, confirming the overlap between recessive and dominant PKD, and two patients with variants in ALG8 and PRKCSH, respectively. Genotype-phenotype correlations showed that patients with PKD1 variants predicted to truncate (T) the protein experienced end-stage renal disease 9 years earlier than patients with PKD1 non-truncating (NT) mutations and >13 years earlier than patients with PKD2 mutations. ADPKD-PKD1 T cases showed a disease onset significantly earlier than ADPKD-PKD1 NT and ADPK-PKD2, as well as a significant earlier diagnosis. These data emphasize the need to combine clinical information with genetic data to achieve useful prognostic predictions.
Keywords: ADPKD; NGS; PKD1; PKD2; cystogenes; polycystic kidney disease.
Copyright © 2020 Mantovani, Bin, Graziano, Capelli, Minardi, Aiello, Ambrosini, Cristalli, Mattiaccio, Pariali, De Fanti, Faletra, Grosso, Cantone, Mancini, Mencarelli, Pasini, Wischmeijer, Sciascia, Seri and La Manna.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous