

Metagenomics reveals impact of geography and acute diarrheal disease on the Central Indian human gut microbiome
- PMID: 32459982
- PMCID: PMC7781581
- DOI: 10.1080/19490976.2020.1752605
Metagenomics reveals impact of geography and acute diarrheal disease on the Central Indian human gut microbiome
Abstract
Background: The Central Indian gut microbiome remains grossly understudied. Herein, we sought to investigate the burden of antimicrobial resistance and diarrheal diseases, particularly Clostridioides difficile, in rural-agricultural and urban populations in Central India, where there is widespread unregulated antibiotic use. We utilized shotgun metagenomics to comprehensively characterize the bacterial and viral fractions of the gut microbiome and their encoded functions in 105 participants.
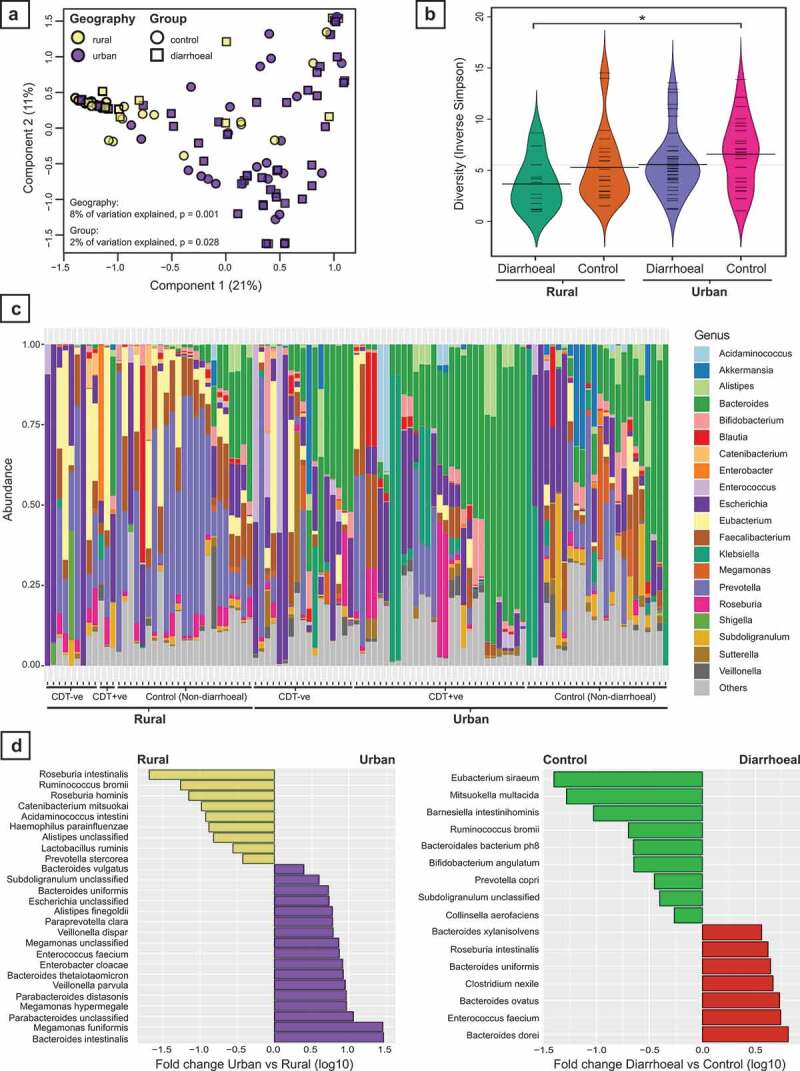
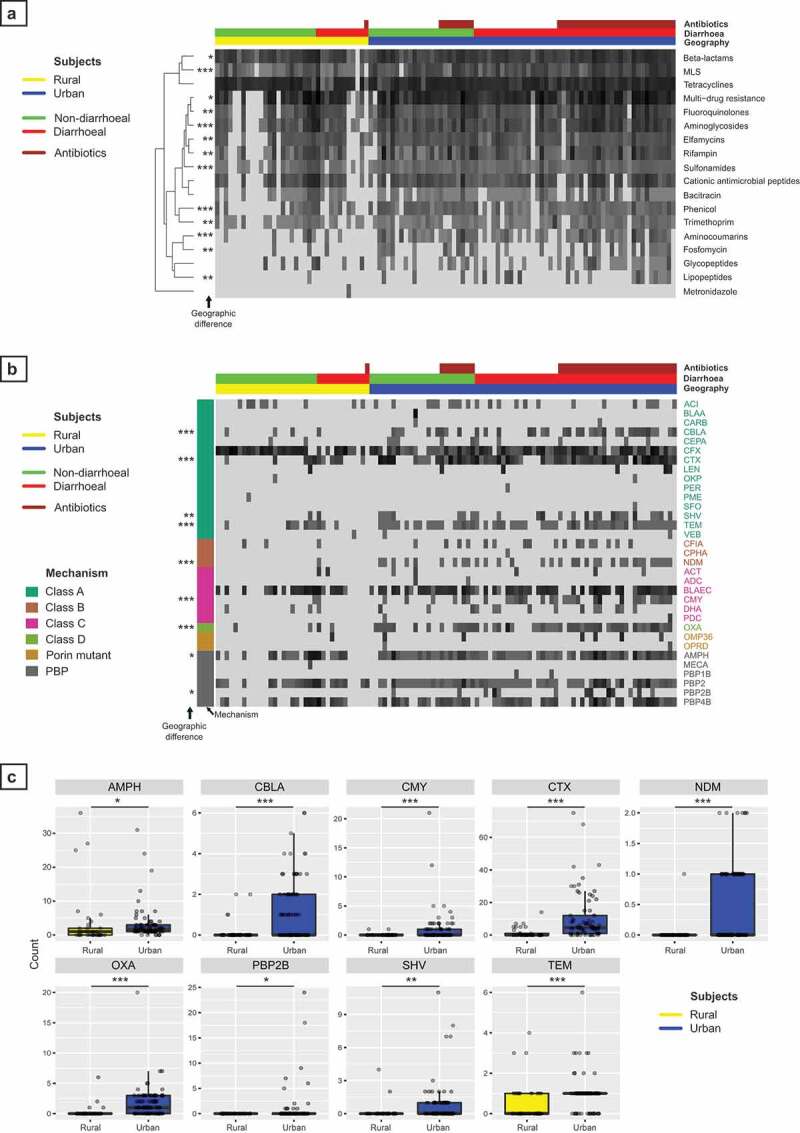
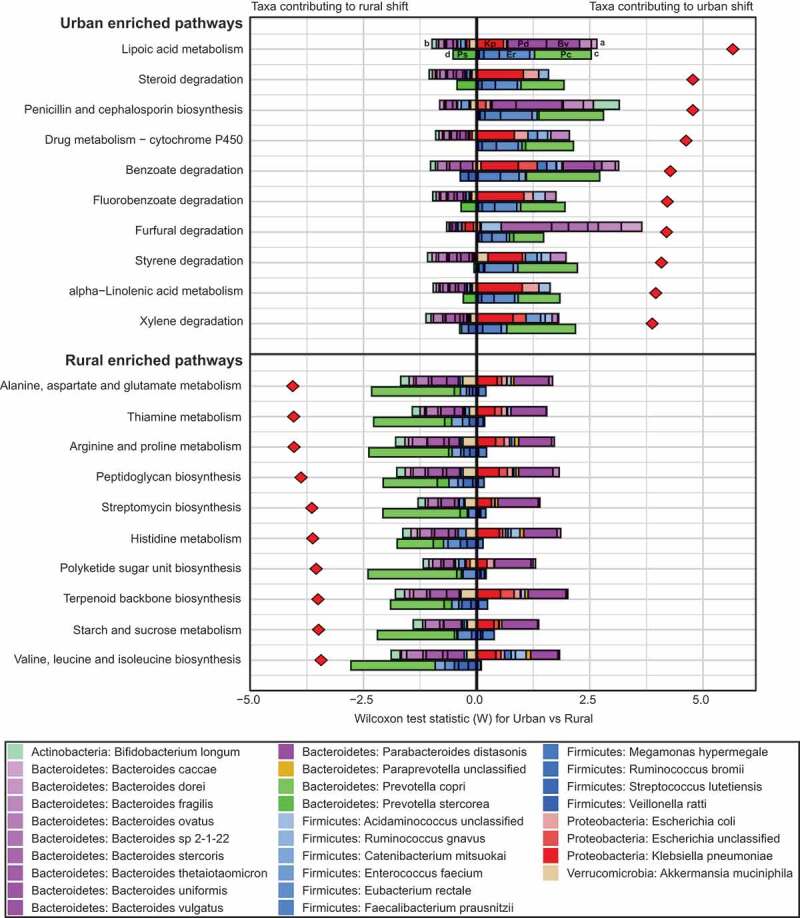
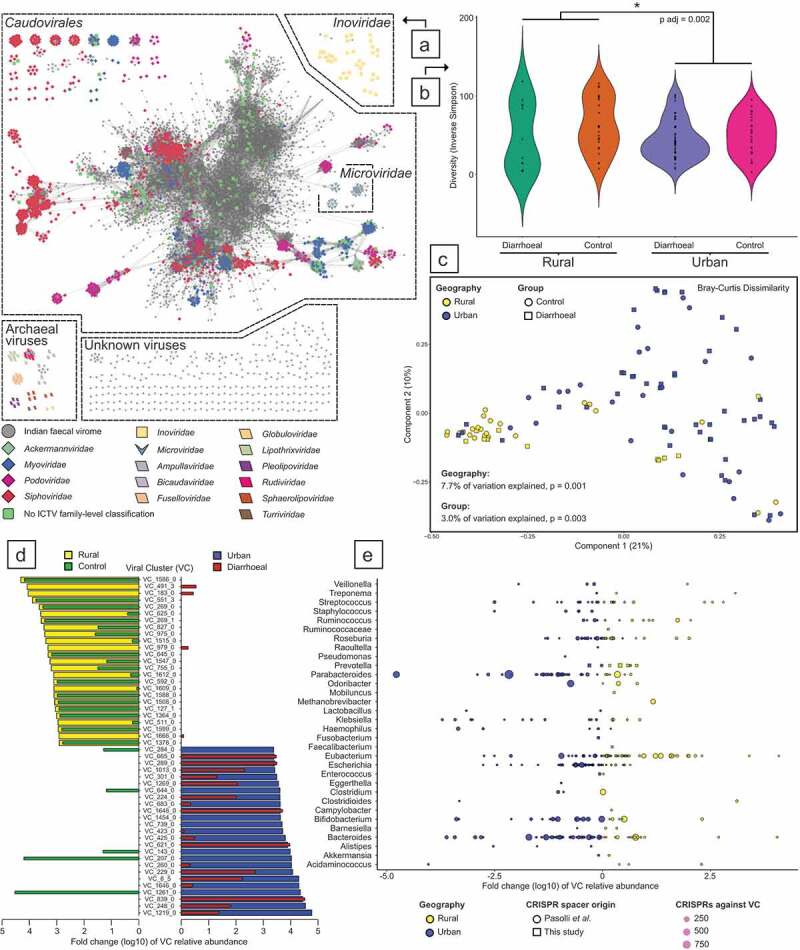
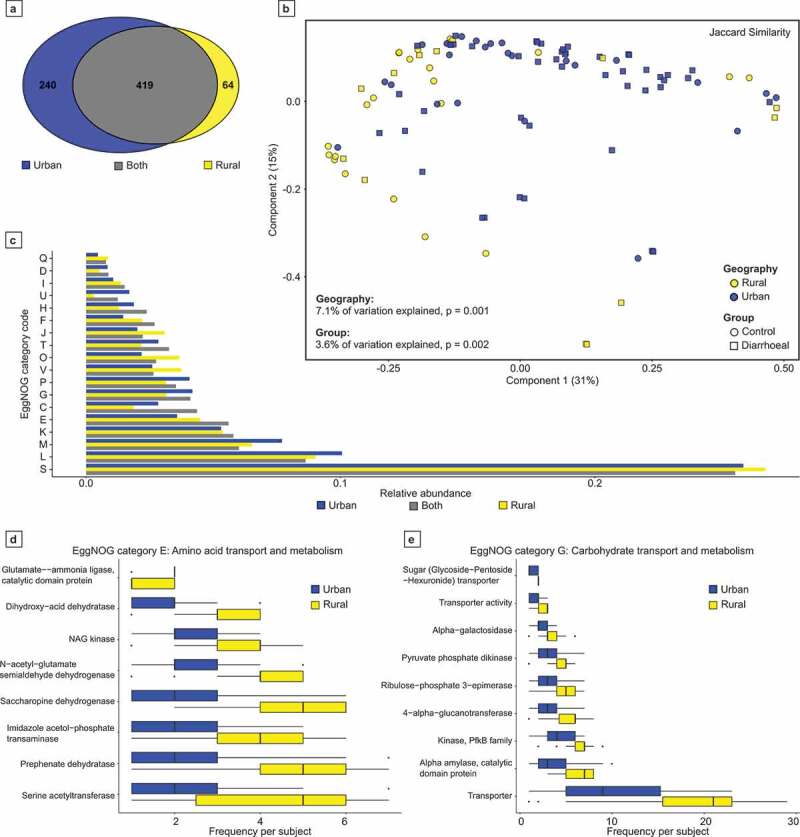
Results: We observed distinct rural-urban differences in bacterial and viral populations, with geography exhibiting a greater influence than diarrheal status. Clostridioides difficile disease was more commonly observed in urban subjects, and their microbiomes were enriched in metabolic pathways relating to the metabolism of industrial compounds and genes encoding resistance to 3rd generation cephalosporins and carbapenems. By linking phages present in the microbiome to their bacterial hosts through CRISPR spacers, phage variation could be directly related to shifts in bacterial populations, with the auxiliary metabolic potential of rural-associated phages enriched for carbon and amino acid energy metabolism.
Conclusions: We report distinct differences in antimicrobial resistance gene profiles, enrichment of metabolic pathways and phage composition between rural and urban populations, as well as a higher burden of Clostridioides difficile disease in the urban population. Our results reveal that geography is the key driver of variation in urban and rural Indian microbiomes, with acute diarrheal disease, including C. difficile disease exerting a lesser impact. Future studies will be required to understand the potential role of dietary, cultural, and genetic factors in contributing to microbiome differences between rural and urban populations.
Keywords: Clostridioides difficile; Central India; Gut microbiome; antibiotic resistome; diarrhea; virome.
Figures
References
-
- Pehrsson EC, Tsukayama P, Patel S, Mejita-Bautista M, Sosa-Soto G, Navarrete KM, Calderon M, Cabrera L, Hoyos-Arango W, Bertoli MT, et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature. 2016;533(7602):212–216. doi:10.1038/nature17672. PMID: 27172044. - DOI - PMC - PubMed
-
- Hendriksen RS, Munk P, Njage P, van Bunnik B, McNally L, Lukjancenko O, Roder T, Nieuwenhuijse D, Pedersen SK, Kjeldgaard J, et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun. 2019;10(1):1124. doi:10.1038/s4167-019-08853-3. PMID: 30850636. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous