

The case of complement activation in COVID-19 multiorgan impact
- PMID: 32461141
- PMCID: PMC7246017
- DOI: 10.1016/j.kint.2020.05.013
The case of complement activation in COVID-19 multiorgan impact
Abstract
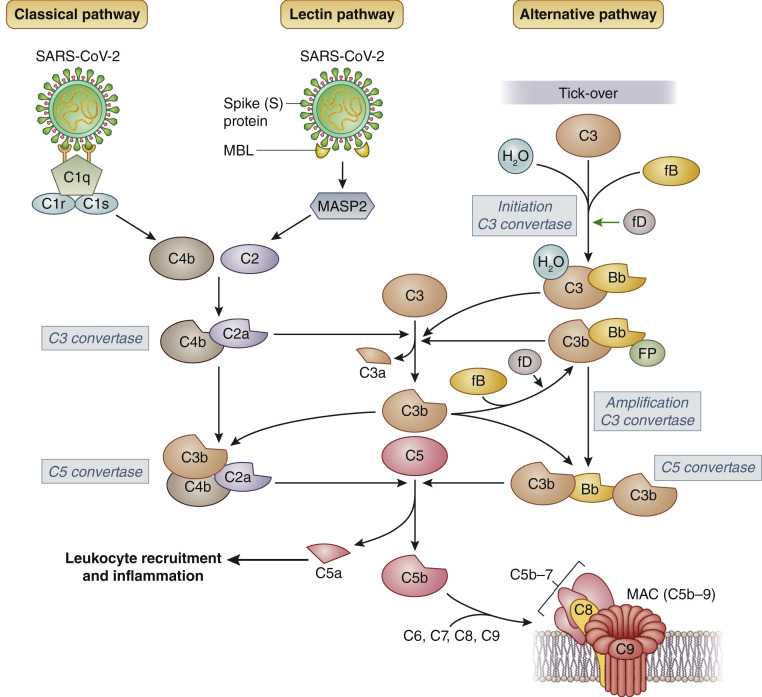
The novel coronavirus disease COVID-19 originates in the lungs, but it may extend to other organs, causing, in severe cases, multiorgan damage, including cardiac injury and acute kidney injury. In severe cases, the presence of kidney injury is associated with increased risk of death, highlighting the relevance of this organ as a target of SARS-CoV-2 infection. COVID-19-associated tissue injury is not primarily mediated by viral infection, but rather is a result of the inflammatory host immune response, which drives hypercytokinemia and aggressive inflammation that affect lung parenchymal cells, diminishing oxygen uptake, but also endothelial cells, resulting in endotheliitis and thrombotic events and intravascular coagulation. The complement system represents the first response of the host immune system to SARS-CoV-2 infection, but there is growing evidence that unrestrained activation of complement induced by the virus in the lungs and other organs plays a major role in acute and chronic inflammation, endothelial cell dysfunction, thrombus formation, and intravascular coagulation, and ultimately contributes to multiple organ failure and death. In this review, we discuss the relative role of the different complement activation products in the pathogenesis of COVID-19-associated tissue inflammation and thrombosis and propose the hypothesis that blockade of the terminal complement pathway may represent a potential therapeutic option for the prevention and treatment of lung and multiorgan damage.
Keywords: C5 inhibition; COVID-19; complement activation; complement terminal pathway; kidney injury; vascular injury.
Copyright © 2020 International Society of Nephrology. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Wadman M., Couzin-Frankel J., Kaiser J., Matacic C. A rampage through the body. Science. 2020;368:356–360. pages 356, 358. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
