

High-resolution annotation of the mouse preimplantation embryo transcriptome using long-read sequencing
- PMID: 32461551
- PMCID: PMC7253418
- DOI: 10.1038/s41467-020-16444-w
High-resolution annotation of the mouse preimplantation embryo transcriptome using long-read sequencing
Erratum in
-
Author Correction: High-resolution annotation of the mouse preimplantation embryo transcriptome using long-read sequencing.Nat Commun. 2021 Mar 15;12(1):1767. doi: 10.1038/s41467-021-22148-6. Nat Commun. 2021. PMID: 33723259 Free PMC article. No abstract available.
Abstract
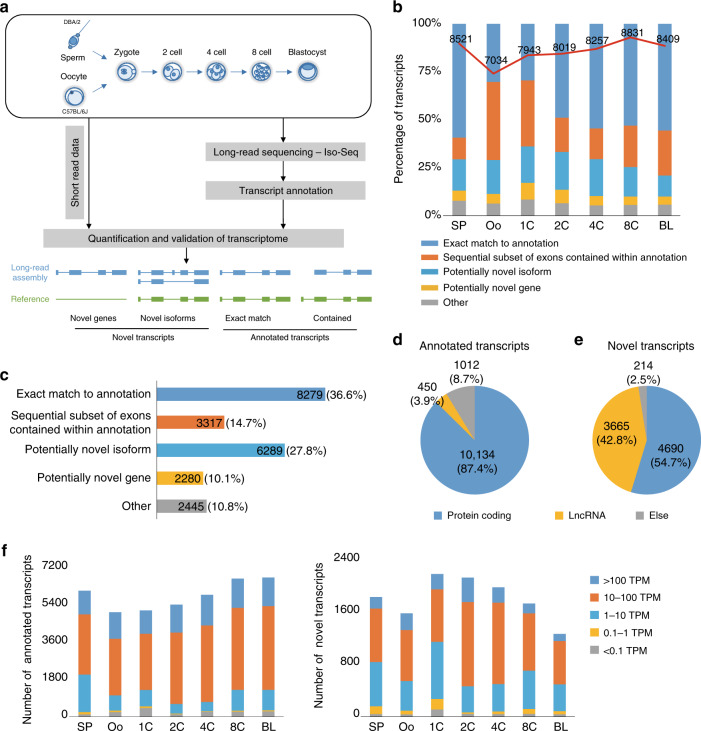
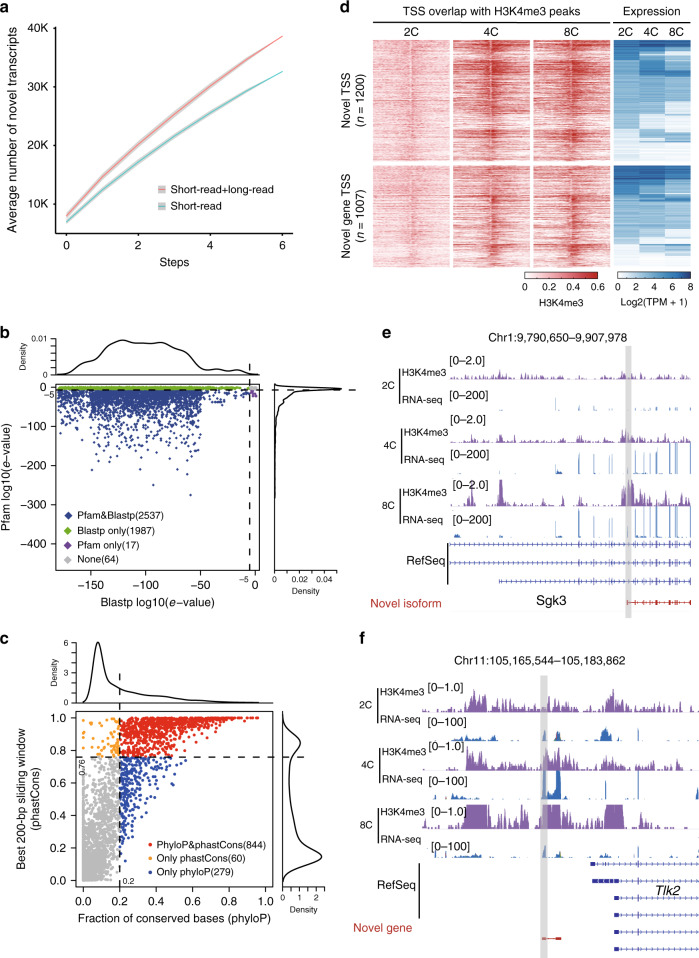
The transcriptome of the preimplantation mouse embryo has been previously annotated by short-read sequencing, with limited coverage and accuracy. Here we utilize a low-cell number transcriptome based on the Smart-seq2 method to perform long-read sequencing. Our analysis describes additional novel transcripts and complexity of the preimplantation transcriptome, identifying 2280 potential novel transcripts from previously unannotated loci and 6289 novel splicing isoforms from previously annotated genes. Notably, these novel transcripts and isoforms with transcription start sites are enriched for an active promoter modification, H3K4me3. Moreover, we generate a more complete and precise transcriptome by combining long-read and short-read data during early embryogenesis. Based on this approach, we identify a previously undescribed isoform of Kdm4dl with a modified mRNA reading frame and a novel noncoding gene designated XLOC_004958. Depletion of Kdm4dl or XLOC_004958 led to abnormal blastocyst development. Thus, our data provide a high-resolution and more precise transcriptome during preimplantation mouse embryogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Mutz KO, Heilkenbrinker A, Lonne M, Walter JG, Stahl F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013;24:22–30. - PubMed
-
- Tang F, et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009;6:377–382. - PubMed
-
- Ke Y, et al. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell. 2017;170:367–381.e320. - PubMed
-
- Liu X, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016;537:558–562. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
