

Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes
- PMID: 32461613
- PMCID: PMC7253413
- DOI: 10.1038/s41467-019-12438-5
Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes
Erratum in
-
Author Correction: Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes.Nat Commun. 2021 Feb 2;12(1):827. doi: 10.1038/s41467-021-21077-8. Nat Commun. 2021. PMID: 33531481 Free PMC article. No abstract available.
Abstract
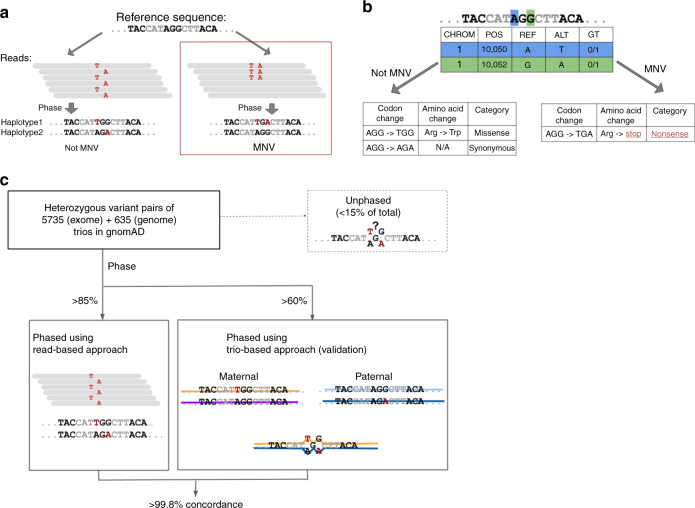
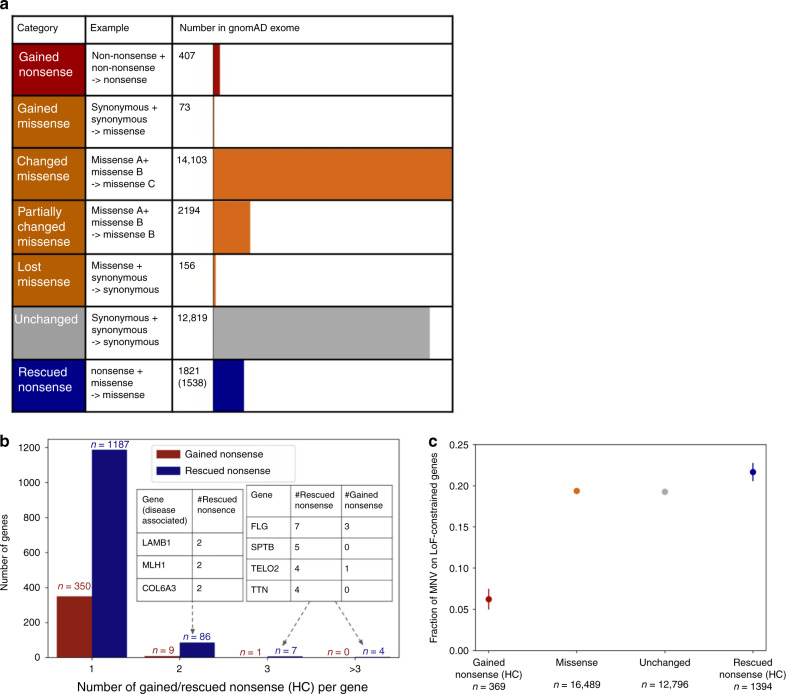
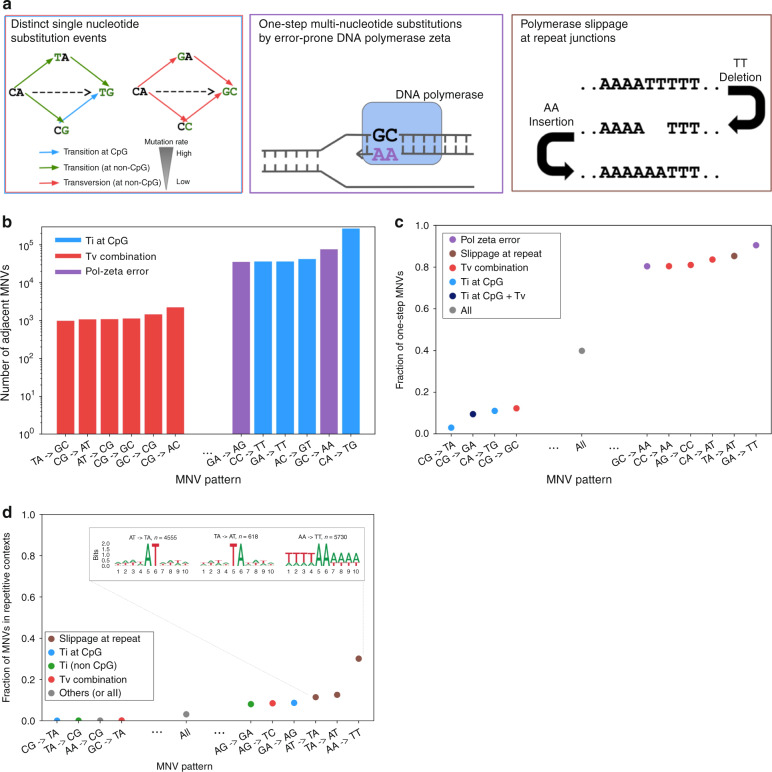
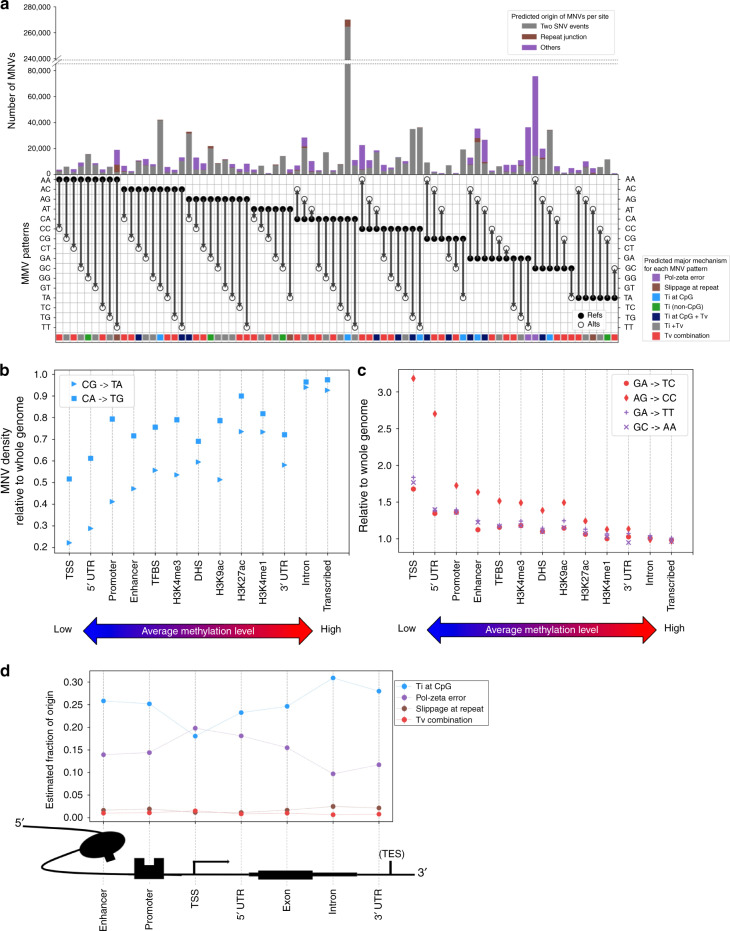
Multi-nucleotide variants (MNVs), defined as two or more nearby variants existing on the same haplotype in an individual, are a clinically and biologically important class of genetic variation. However, existing tools typically do not accurately classify MNVs, and understanding of their mutational origins remains limited. Here, we systematically survey MNVs in 125,748 whole exomes and 15,708 whole genomes from the Genome Aggregation Database (gnomAD). We identify 1,792,248 MNVs across the genome with constituent variants falling within 2 bp distance of one another, including 18,756 variants with a novel combined effect on protein sequence. Finally, we estimate the relative impact of known mutational mechanisms - CpG deamination, replication error by polymerase zeta, and polymerase slippage at repeat junctions - on the generation of MNVs. Our results demonstrate the value of haplotype-aware variant annotation, and refine our understanding of genome-wide mutational mechanisms of MNVs.
Conflict of interest statement
D.G.M. is a founder with equity in Goldfinch Bio, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi-Genzyme. K.J.K. owns stock in Personalis. E.V.M. has received research support in the form of charitable contributions from Charles River Laboratories and Ionis Pharmaceuticals, and has consulted for Deerfield Management. M.I.M.: The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. He has served on advisory panels for Pfizer, NovoNordisk, Zoe Global; has received honoraria from Merck, Pfizer, NovoNordisk, and Eli Lilly; has stock options in Zoe Global and has received research funding from Abbvie, Astra Zeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, NovoNordisk, Pfizer, Roche, Sanofi Aventis, Servier, and Takeda. As of June 2019, M.I.M. is an employee of Genentech, and holds stock in Roche. R.K.W. has received unrestricted research grants from Takeda Pharmaceutical Company. M.J.D. is a founder of Maze Therapeutics. B.M.N. is a member of the scientific advisory board at Deep Genomics and consultant for Camp4 Therapeutics, Takeda Pharmaceutical, and Biogen. A.O.D.L. has received honoraria from ARUP and Chan Zuckerberg Initiative.
Figures
Comment in
-
Exploring human genomic diversity with gnomAD.Nat Rev Genet. 2020 Aug;21(8):448. doi: 10.1038/s41576-020-0255-7. Nat Rev Genet. 2020. PMID: 32488197 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
