

Characterising the loss-of-function impact of 5' untranslated region variants in 15,708 individuals
- PMID: 32461616
- PMCID: PMC7253449
- DOI: 10.1038/s41467-019-10717-9
Characterising the loss-of-function impact of 5' untranslated region variants in 15,708 individuals
Erratum in
-
Author Correction: Characterising the loss-of-function impact of 5' untranslated region variants in 15,708 individuals.Nat Commun. 2021 Feb 2;12(1):839. doi: 10.1038/s41467-021-21052-3. Nat Commun. 2021. PMID: 33531501 Free PMC article. No abstract available.
Abstract
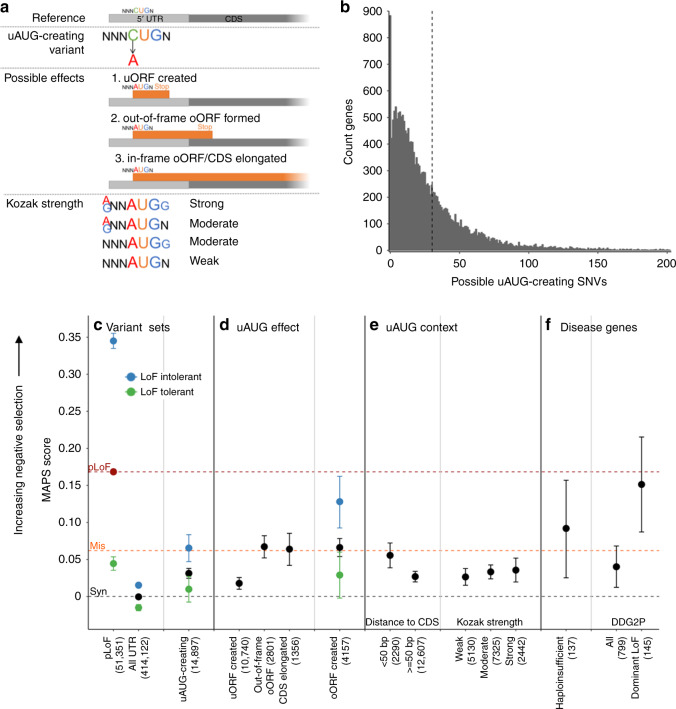
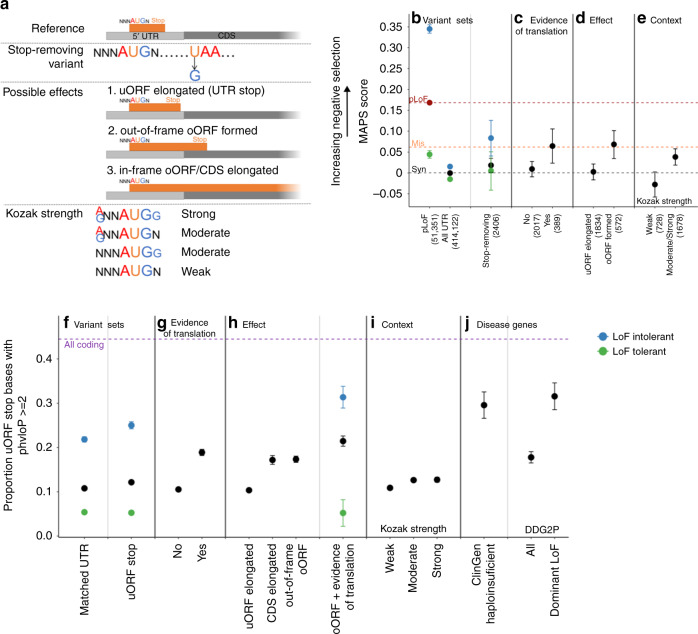
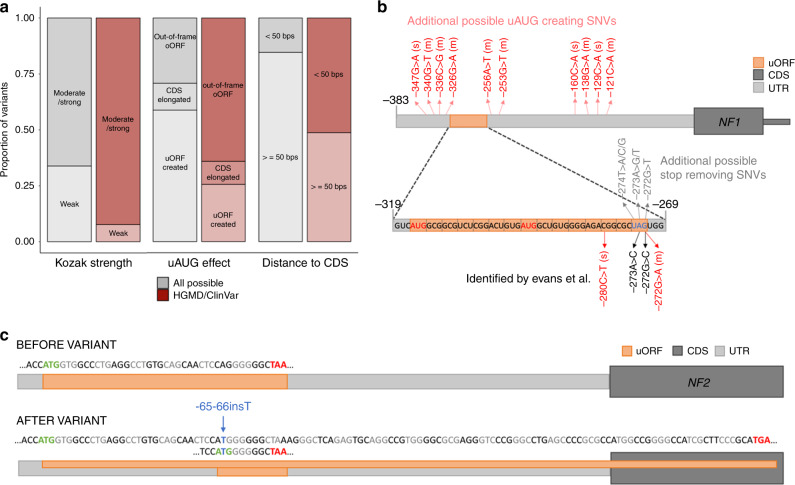
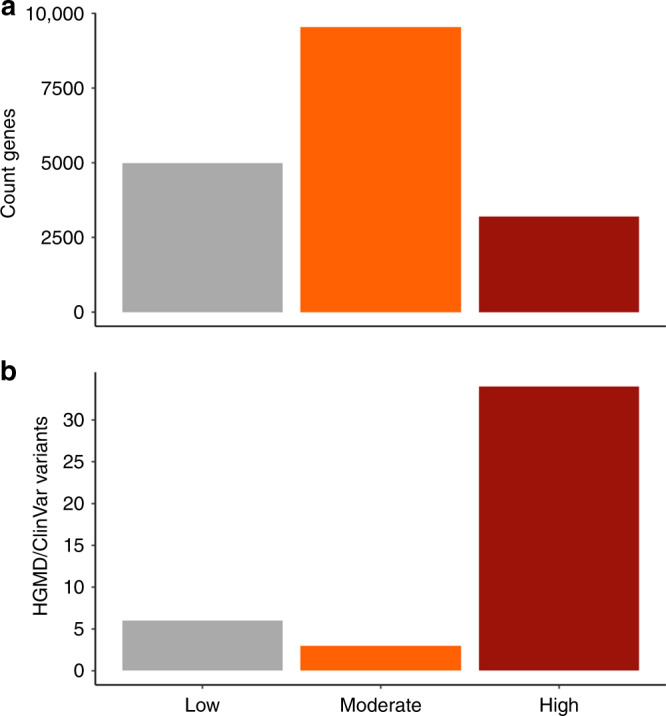
Upstream open reading frames (uORFs) are tissue-specific cis-regulators of protein translation. Isolated reports have shown that variants that create or disrupt uORFs can cause disease. Here, in a systematic genome-wide study using 15,708 whole genome sequences, we show that variants that create new upstream start codons, and variants disrupting stop sites of existing uORFs, are under strong negative selection. This selection signal is significantly stronger for variants arising upstream of genes intolerant to loss-of-function variants. Furthermore, variants creating uORFs that overlap the coding sequence show signals of selection equivalent to coding missense variants. Finally, we identify specific genes where modification of uORFs likely represents an important disease mechanism, and report a novel uORF frameshift variant upstream of NF2 in neurofibromatosis. Our results highlight uORF-perturbing variants as an under-recognised functional class that contribute to penetrant human disease, and demonstrate the power of large-scale population sequencing data in studying non-coding variant classes.
Conflict of interest statement
D.G.M. is a founder with equity in Goldfinch Bio. The remaining authors declare no competing interests.
Figures
Comment in
-
Exploring human genomic diversity with gnomAD.Nat Rev Genet. 2020 Aug;21(8):448. doi: 10.1038/s41576-020-0255-7. Nat Rev Genet. 2020. PMID: 32488197 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- MC_U120085815/MRC_/Medical Research Council/United Kingdom
- HICF-R6-373/DH_/Department of Health/United Kingdom
- CS/14/2/30841/BHF_/British Heart Foundation/United Kingdom
- 107469/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- MC_UP_1102/20/MRC_/Medical Research Council/United Kingdom
- R01 MH085548/MH/NIMH NIH HHS/United States
- IS-BRC-1215-20007/DH_/Department of Health/United Kingdom
- R01 MH123451/MH/NIMH NIH HHS/United States
- U54 DK105566/DK/NIDDK NIH HHS/United States
- R01 GM104371/GM/NIGMS NIH HHS/United States
- RG/18/10/33842/BHF_/British Heart Foundation/United Kingdom
- P30 DK020572/DK/NIDDK NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
