

Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell
- PMID: 32463360
- PMCID: PMC7255802
- DOI: 10.7554/eLife.49178
Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell
Abstract
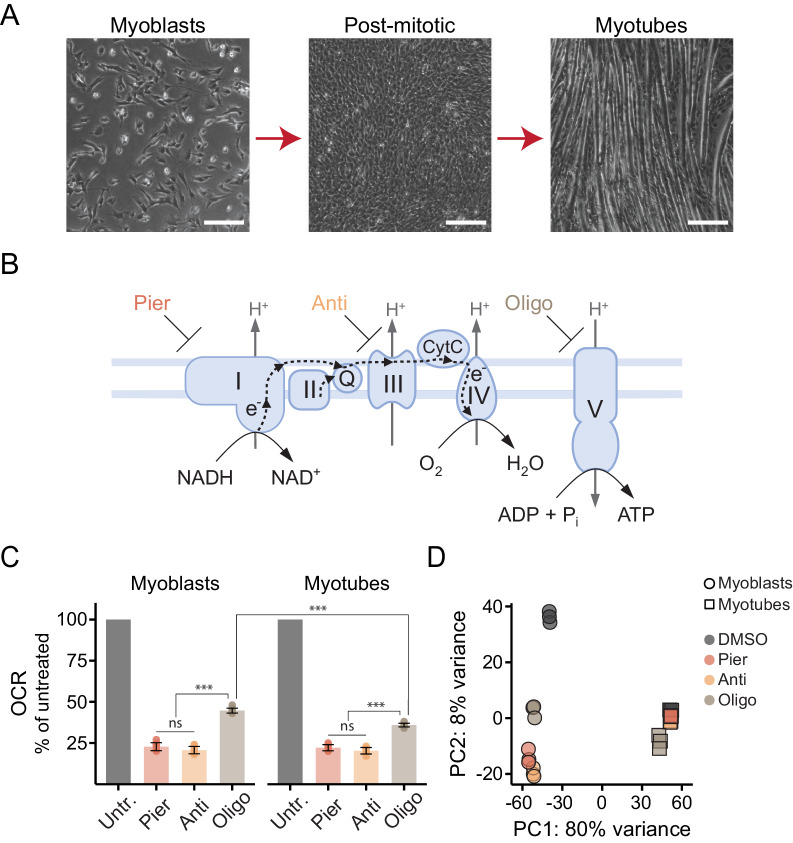
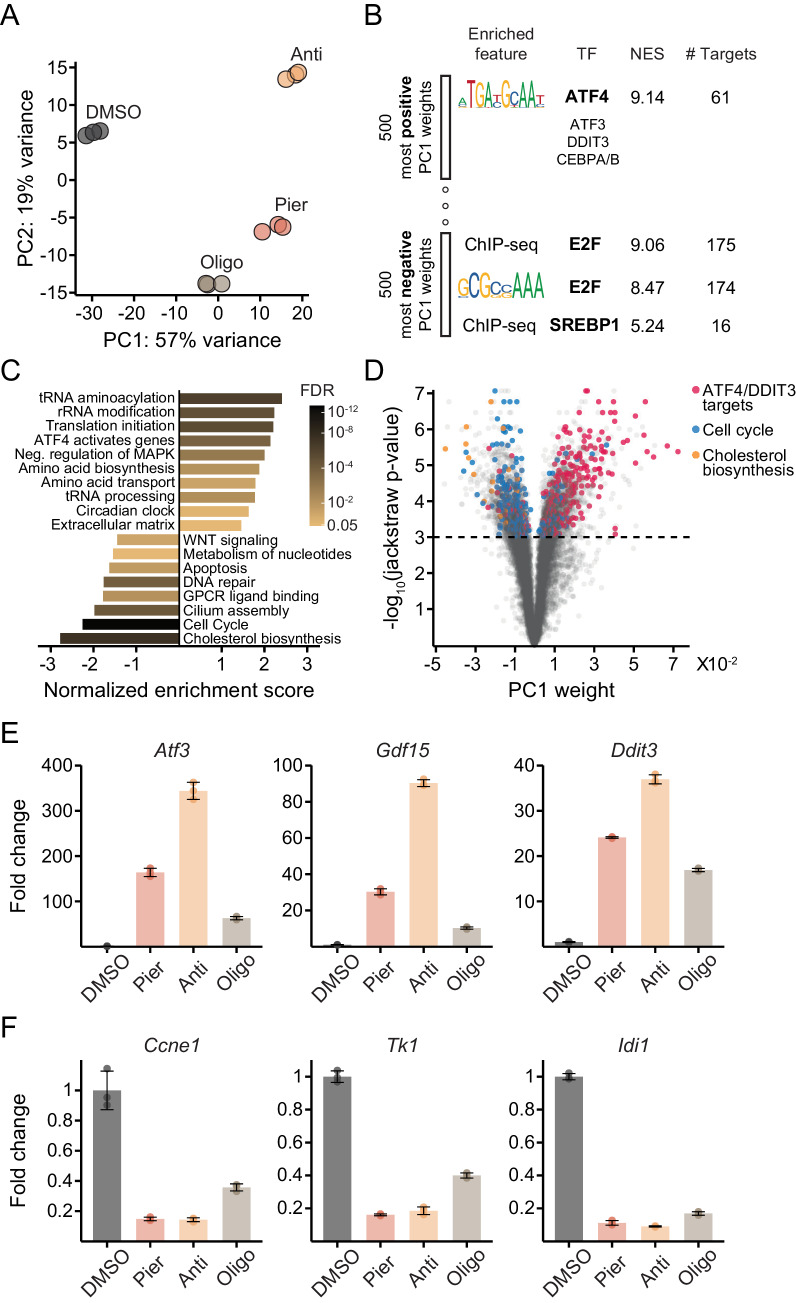
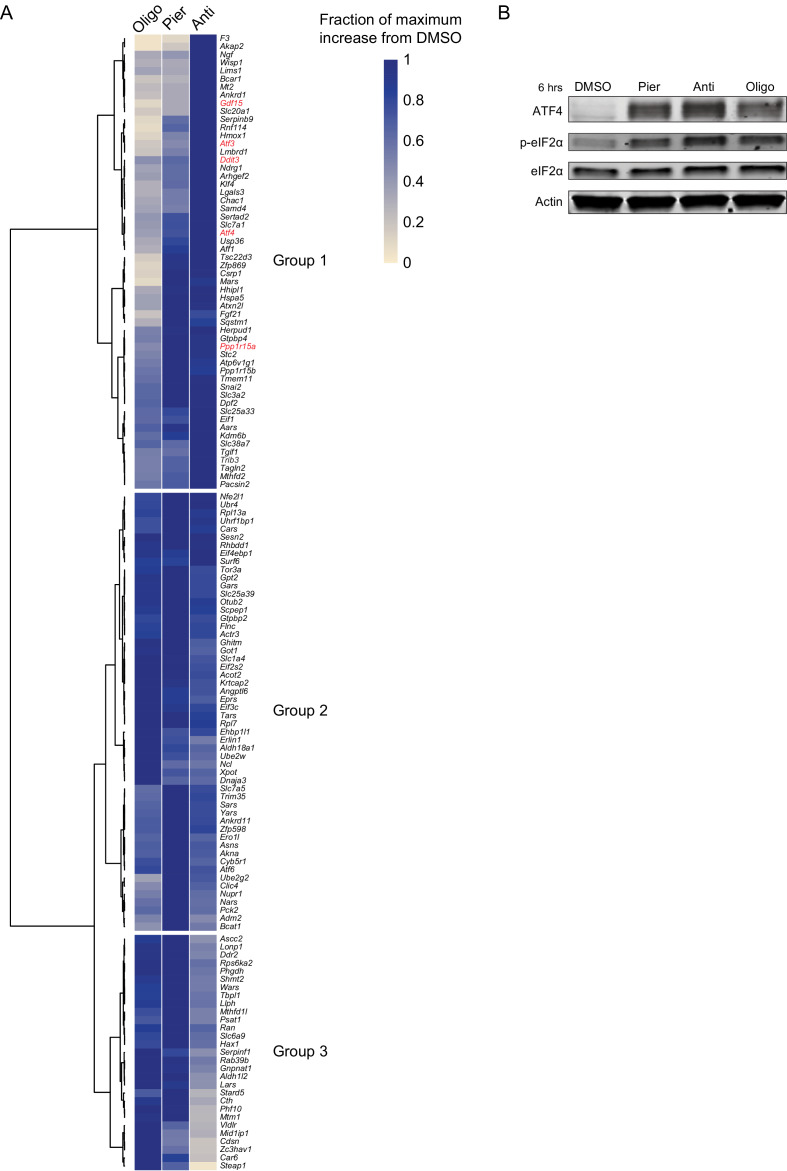
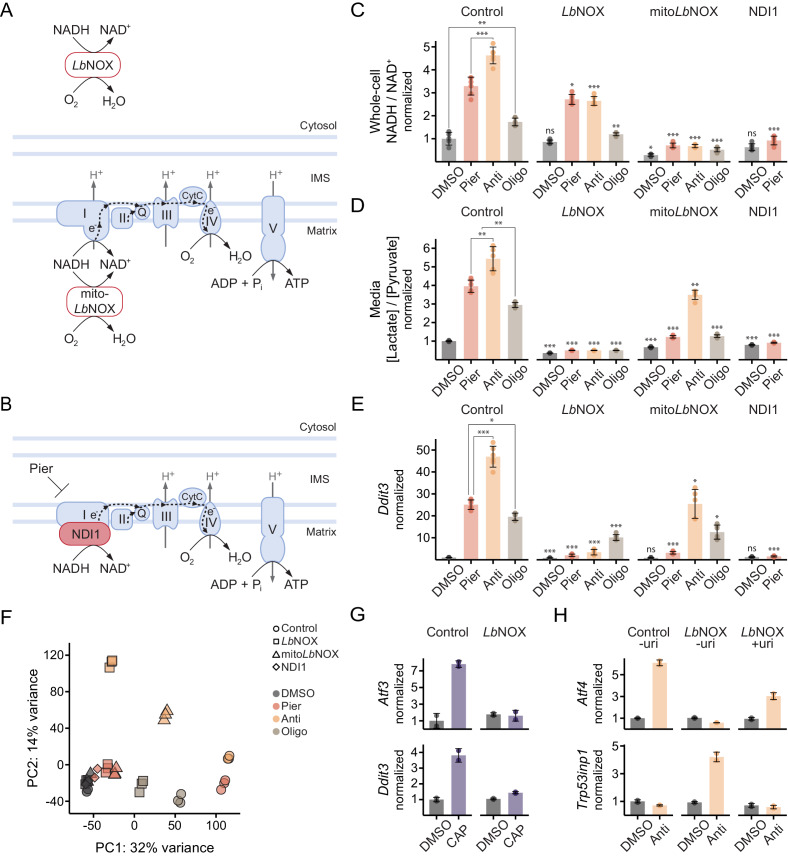
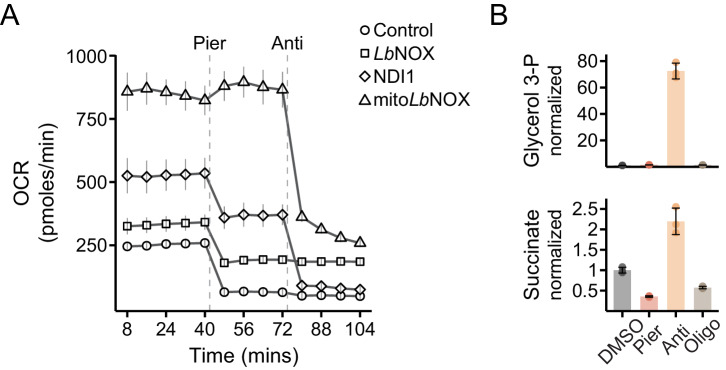
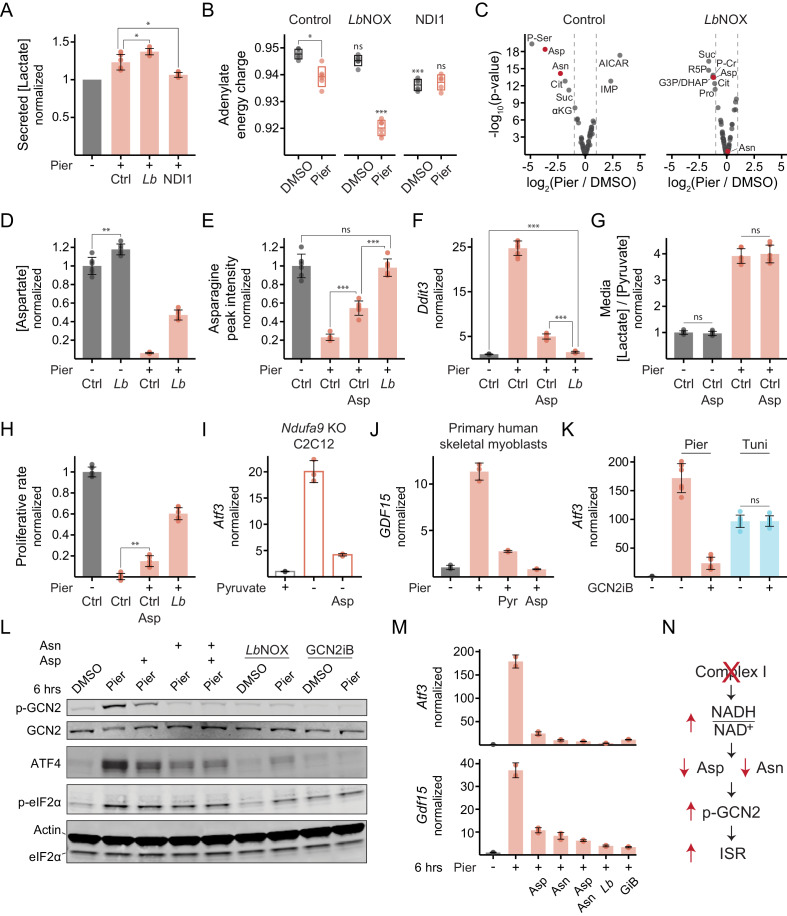
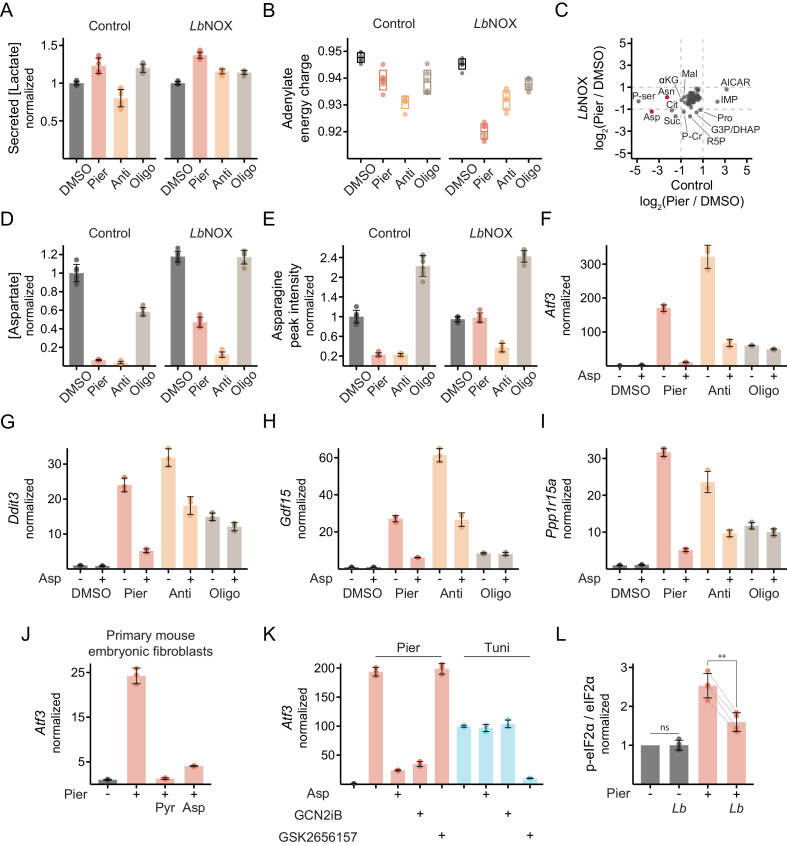
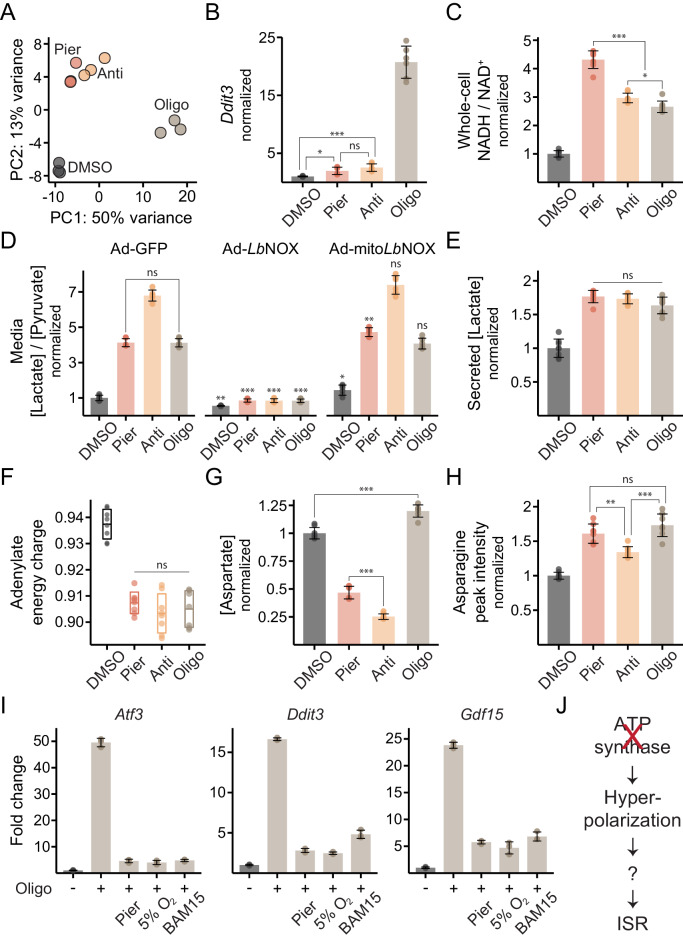
Mitochondrial dysfunction is associated with activation of the integrated stress response (ISR) but the underlying triggers remain unclear. We systematically combined acute mitochondrial inhibitors with genetic tools for compartment-specific NADH oxidation to trace mechanisms linking different forms of mitochondrial dysfunction to the ISR in proliferating mouse myoblasts and in differentiated myotubes. In myoblasts, we find that impaired NADH oxidation upon electron transport chain (ETC) inhibition depletes asparagine, activating the ISR via the eIF2α kinase GCN2. In myotubes, however, impaired NADH oxidation following ETC inhibition neither depletes asparagine nor activates the ISR, reflecting an altered metabolic state. ATP synthase inhibition in myotubes triggers the ISR via a distinct mechanism related to mitochondrial inner-membrane hyperpolarization. Our work dispels the notion of a universal path linking mitochondrial dysfunction to the ISR, instead revealing multiple paths that depend both on the nature of the mitochondrial defect and on the metabolic state of the cell.
Keywords: ATF4; GCN2; genetics; genomics; human; human biology; integrated stress response; medicine; metabolism; mitochondria; mouse; p53.
© 2020, Mick et al.
Conflict of interest statement
EM listed as an inventor on a patent filed by Massachusetts General Hospital on the therapeutic uses of GCN2 inhibitors. DT, RS, AJ No competing interests declared, OS a paid consultant for Proteinaceous Inc. VM listed as an inventor on a patent filed by Massachusetts General Hospital on the therapeutic uses of GCN2 inhibitors, and a paid advisor to Janssen Pharmaceuticals and 5am Ventures.
Figures

References
-
- Alfadda A, DosSantos RA, Stepanyan Z, Marrif H, Silva JE. Mice with deletion of the mitochondrial glycerol-3-phosphate dehydrogenase gene exhibit a thrifty phenotype: effect of gender. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2004;287:R147–R156. doi: 10.1152/ajpregu.00103.2004. - DOI - PubMed
-
- Andrulis IL, Chiang CS, Arfin SM, Miner TA, Hatfield GW. Biochemical characterization of a mutant asparaginyl-tRNA synthetase from chinese hamster ovary cells. The Journal of Biological Chemistry. 1978;253:58–62. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
