

Improved bacterial recombineering by parallelized protein discovery
- PMID: 32467157
- PMCID: PMC7306799
- DOI: 10.1073/pnas.2001588117
Improved bacterial recombineering by parallelized protein discovery
Abstract
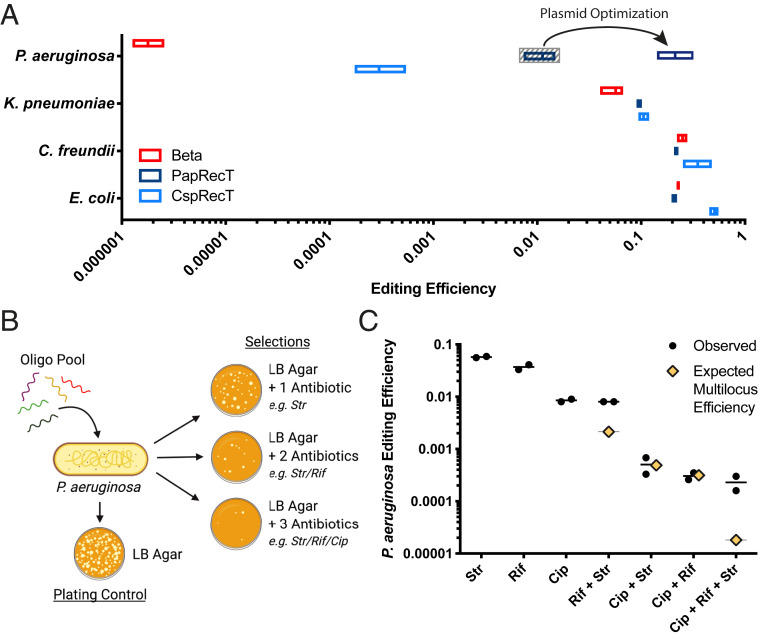
Exploiting bacteriophage-derived homologous recombination processes has enabled precise, multiplex editing of microbial genomes and the construction of billions of customized genetic variants in a single day. The techniques that enable this, multiplex automated genome engineering (MAGE) and directed evolution with random genomic mutations (DIvERGE), are however, currently limited to a handful of microorganisms for which single-stranded DNA-annealing proteins (SSAPs) that promote efficient recombineering have been identified. Thus, to enable genome-scale engineering in new hosts, efficient SSAPs must first be found. Here we introduce a high-throughput method for SSAP discovery that we call "serial enrichment for efficient recombineering" (SEER). By performing SEER in Escherichia coli to screen hundreds of putative SSAPs, we identify highly active variants PapRecT and CspRecT. CspRecT increases the efficiency of single-locus editing to as high as 50% and improves multiplex editing by 5- to 10-fold in E. coli, while PapRecT enables efficient recombineering in Pseudomonas aeruginosa, a concerning human pathogen. CspRecT and PapRecT are also active in other, clinically and biotechnologically relevant enterobacteria. We envision that the deployment of SEER in new species will pave the way toward pooled interrogation of genotype-to-phenotype relationships in previously intractable bacteria.
Keywords: MAGE; RecT; genome engineering; recombineering; synthetic biology.
Conflict of interest statement
Competing interest statement: G.M.C. has related financial interests in EnEvolv, GRO Biosciences, and 64-x. G.M.C., C.J.G., M.J.L., and X.R. have submitted a patent application relating to pieces of this work (WO2017184227A2). T.M.W., G.T.F., and G.M.C. have submitted a patent application related to the improved single-stranded DNA-annealing proteins variants referenced here. A.N. and C.P. have submitted a patent application related to directed evolution with random genomic mutations (DIvERGE) (PCT/EP2017/082574 [WO2018108987] Mutagenizing Intracellular Nucleic Acids).
Figures



References
-
- Bird A. W. et al. ., High-efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes. Nat. Methods 9, 103–109 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
