

Amyloidogenic processing of amyloid β protein precursor (APP) is enhanced in the brains of alcadein α-deficient mice
- PMID: 32467230
- PMCID: PMC7363152
- DOI: 10.1074/jbc.RA119.012386
Amyloidogenic processing of amyloid β protein precursor (APP) is enhanced in the brains of alcadein α-deficient mice
Abstract
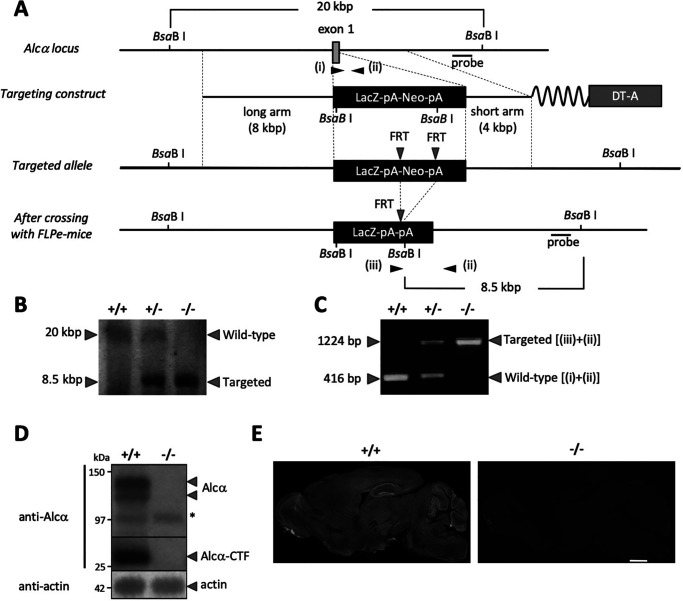
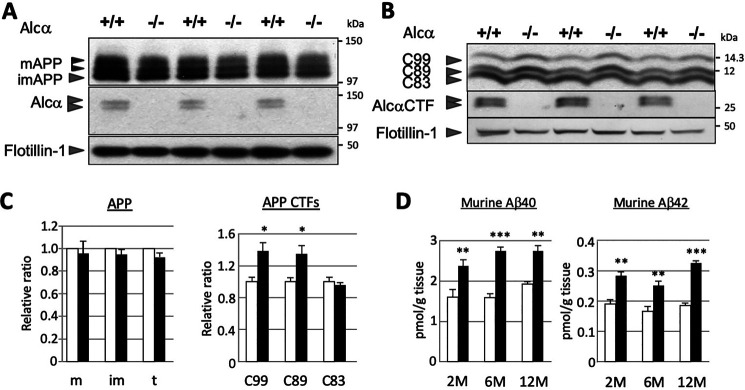
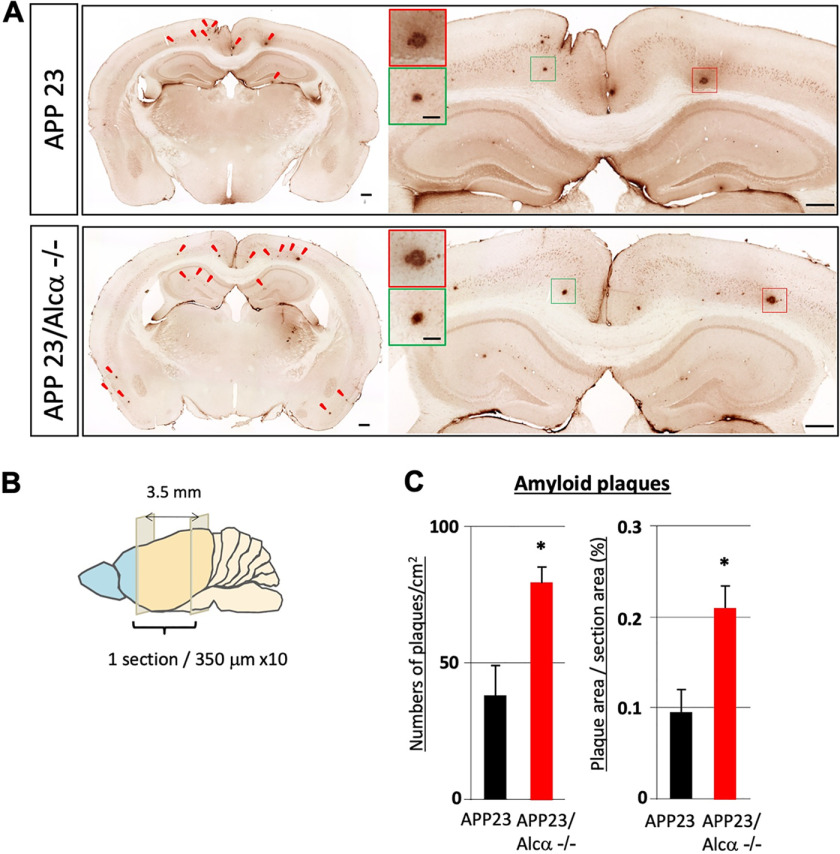
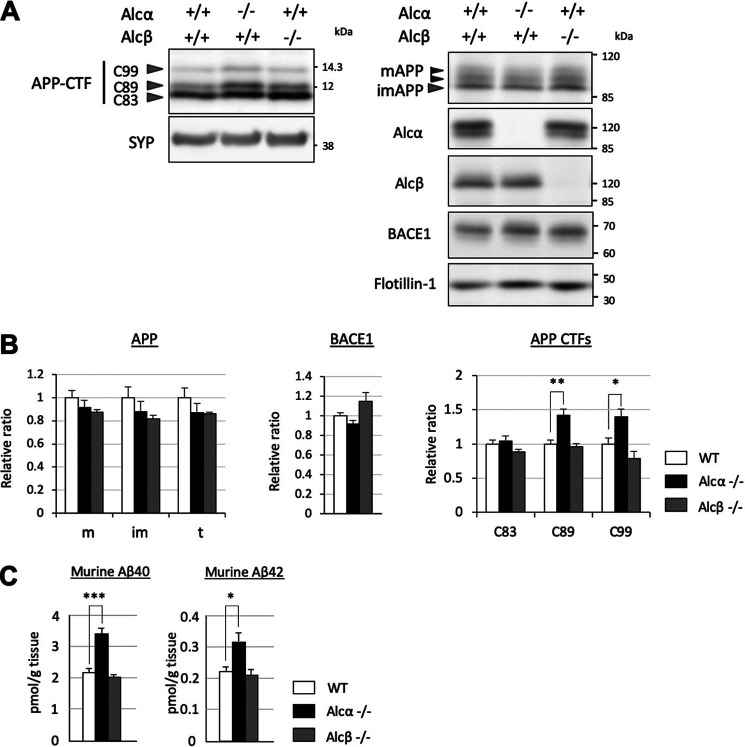
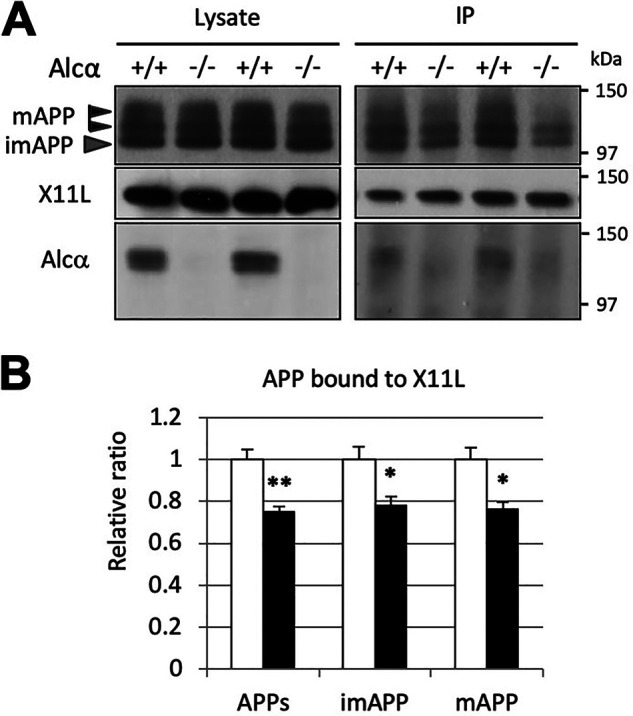
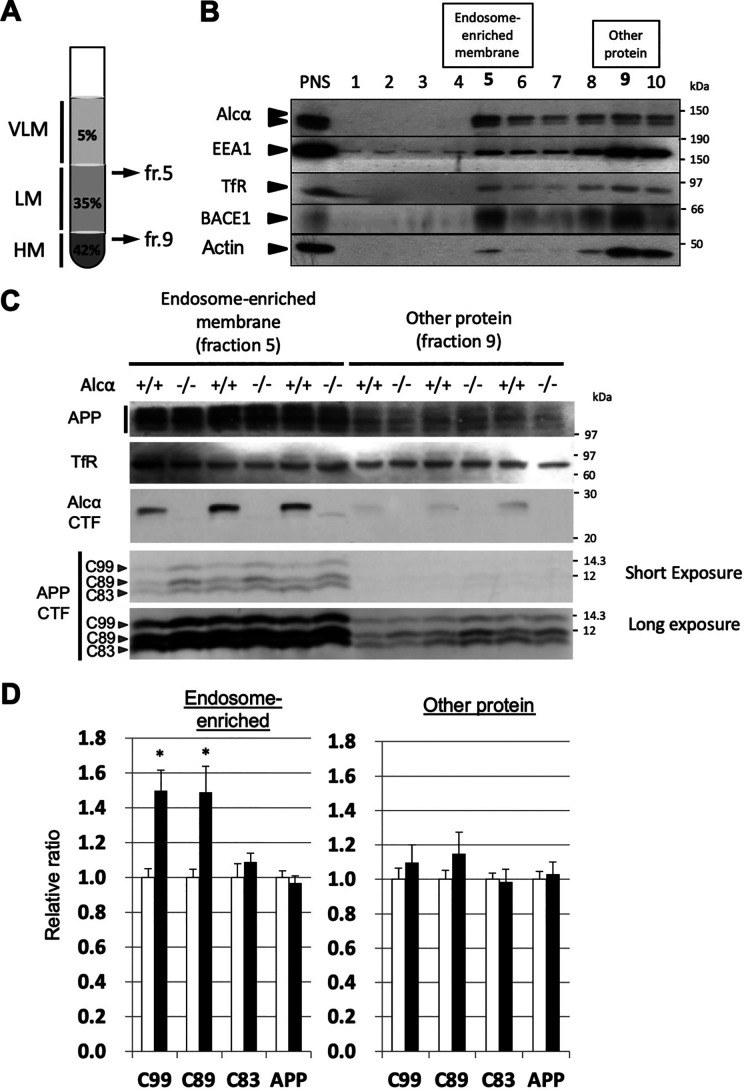
Alzheimer's disease (AD) is a very common neurodegenerative disorder, chiefly caused by increased production of neurotoxic β-amyloid (Aβ) peptide generated from proteolytic cleavage of β-amyloid protein precursor (APP). Except for familial AD arising from mutations in the APP and presenilin (PSEN) genes, the molecular mechanisms regulating the amyloidogenic processing of APP are largely unclear. Alcadein α/calsyntenin1 (ALCα/CLSTN1) is a neuronal type I transmembrane protein that forms a complex with APP, mediated by the neuronal adaptor protein X11-like (X11L or MINT2). Formation of the ALCα-X11L-APP tripartite complex suppresses Aβ generation in vitro, and X11L-deficient mice exhibit enhanced amyloidogenic processing of endogenous APP. However, the role of ALCα in APP metabolism in vivo remains unclear. Here, by generating ALCα-deficient mice and using immunohistochemistry, immunoblotting, and co-immunoprecipitation analyses, we verified the role of ALCα in the suppression of amyloidogenic processing of endogenous APP in vivo We observed that ALCα deficiency attenuates the association of X11L with APP, significantly enhances amyloidogenic β-site cleavage of APP, especially in endosomes, and increases the generation of endogenous Aβ in the brain. Furthermore, we noted amyloid plaque formation in the brains of human APP-transgenic mice in an ALCα-deficient background. These results unveil a potential role of ALCα in protecting cerebral neurons from Aβ-dependent pathogenicity in AD.
Keywords: Alzheimer's disease; Mint2; X11-like; alcadein; amyloid β (Aβ); amyloid β protein precursor (APP); calsyntenin; membrane protein; metabolism; neurodegeneration.
© 2020 Gotoh et al.
Conflict of interest statement
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
Suppression of the amyloidogenic metabolism of APP and the accumulation of Aβ by alcadein α in the brain during aging.Sci Rep. 2024 Aug 9;14(1):18471. doi: 10.1038/s41598-024-69400-9. Sci Rep. 2024. PMID: 39122814 Free PMC article.
-
X11 proteins regulate the translocation of amyloid beta-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by beta-site-cleaving enzyme in brain.J Biol Chem. 2008 Dec 19;283(51):35763-71. doi: 10.1074/jbc.M801353200. Epub 2008 Oct 9. J Biol Chem. 2008. PMID: 18845544 Free PMC article.
-
Cytoplasmic fragment of Alcadein α generated by regulated intramembrane proteolysis enhances amyloid β-protein precursor (APP) transport into the late secretory pathway and facilitates APP cleavage.J Biol Chem. 2015 Jan 9;290(2):987-95. doi: 10.1074/jbc.M114.599852. Epub 2014 Nov 18. J Biol Chem. 2015. PMID: 25406318 Free PMC article.
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
-
Proteolytic processing of Alzheimer's β-amyloid precursor protein.J Neurochem. 2012 Jan;120 Suppl 1(Suppl 1):9-21. doi: 10.1111/j.1471-4159.2011.07519.x. Epub 2011 Nov 28. J Neurochem. 2012. PMID: 22122372 Free PMC article. Review.
Cited by
-
Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer's disease and normal physiology.Front Cell Dev Biol. 2022 Jul 26;10:969547. doi: 10.3389/fcell.2022.969547. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35959488 Free PMC article. Review.
-
Suppression of the amyloidogenic metabolism of APP and the accumulation of Aβ by alcadein α in the brain during aging.Sci Rep. 2024 Aug 9;14(1):18471. doi: 10.1038/s41598-024-69400-9. Sci Rep. 2024. PMID: 39122814 Free PMC article.
-
Zinc in Cognitive Impairment and Aging.Biomolecules. 2022 Jul 18;12(7):1000. doi: 10.3390/biom12071000. Biomolecules. 2022. PMID: 35883555 Free PMC article. Review.
-
Brain p3-Alcβ peptide restores neuronal viability impaired by Alzheimer's amyloid β-peptide.EMBO Mol Med. 2023 May 8;15(5):e17052. doi: 10.15252/emmm.202217052. Epub 2023 Mar 30. EMBO Mol Med. 2023. PMID: 36994913 Free PMC article.
-
Unveiling New Genetic Variants Associated with Age at Onset in Alzheimer's Disease and Frontotemporal Lobar Degeneration Due to C9orf72 Repeat Expansions.Int J Mol Sci. 2024 Jul 7;25(13):7457. doi: 10.3390/ijms25137457. Int J Mol Sci. 2024. PMID: 39000564 Free PMC article.
References
-
- Kakuda N., Shoji M., Arai H., Furukawa K., Ikeuchi T., Akazawa K., Takami M., Hatsuta H., Murayama S., Hashimoto Y., Miyajima M., Arai H., Nagashima Y., Yamaguchi H., Kuwano R., et al. (2012) Altered γ-secretase activity in mild cognitive impairment and Alzheimer's disease. EMBO Mol. Med. 4, 344–352 10.1002/emmm.201200214 - DOI - PMC - PubMed
-
- Goate A., Chartier-Harlin M.-C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L., Mant R., Newton P., Rooke K., Roques P., Talbot C., et al. (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706 10.1038/349704a0 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
