

Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution
- PMID: 32469868
- PMCID: PMC7286533
- DOI: 10.1371/journal.pgen.1008827
Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution
Abstract
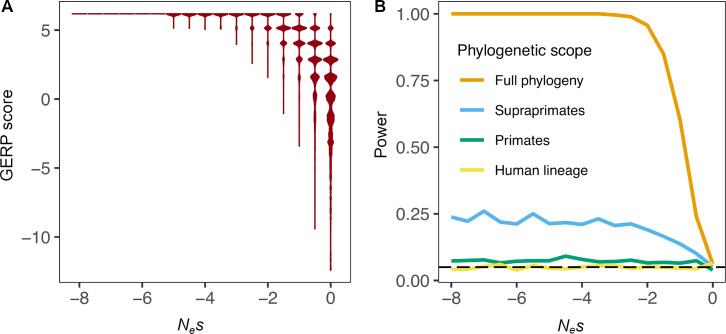
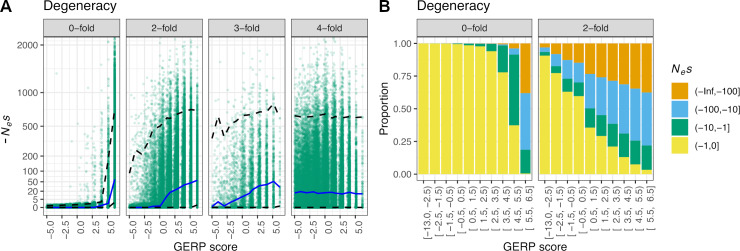
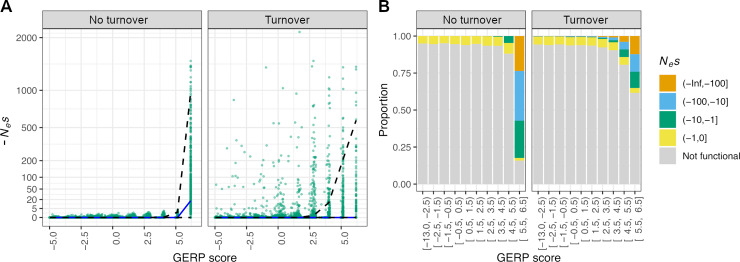
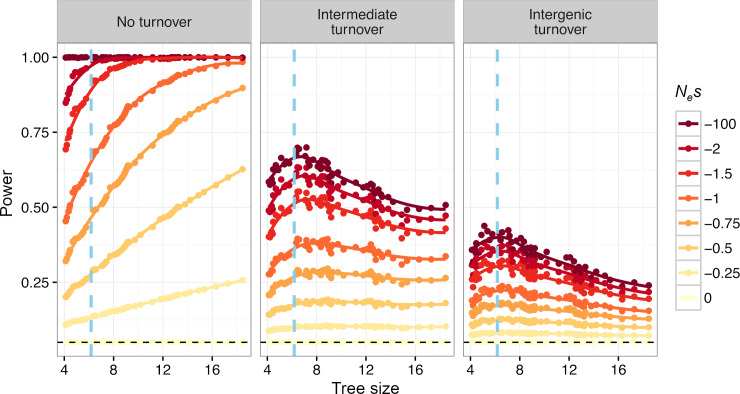
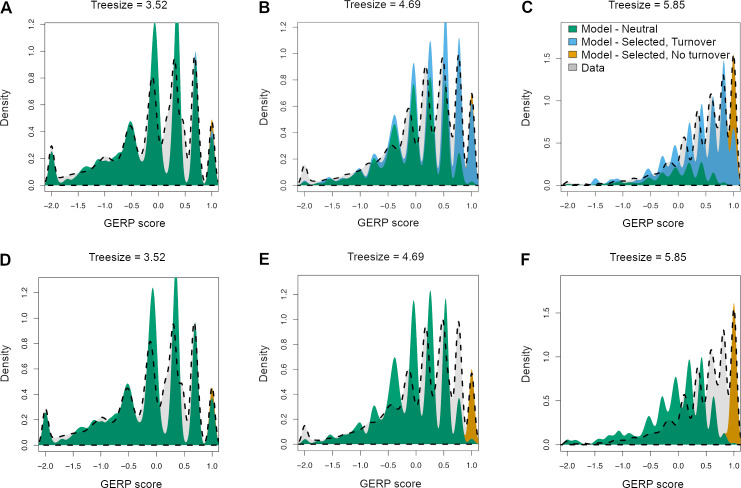
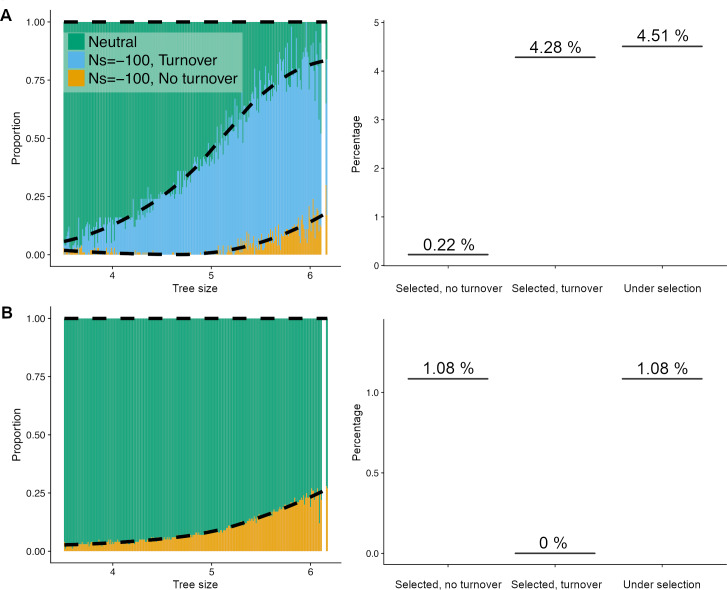
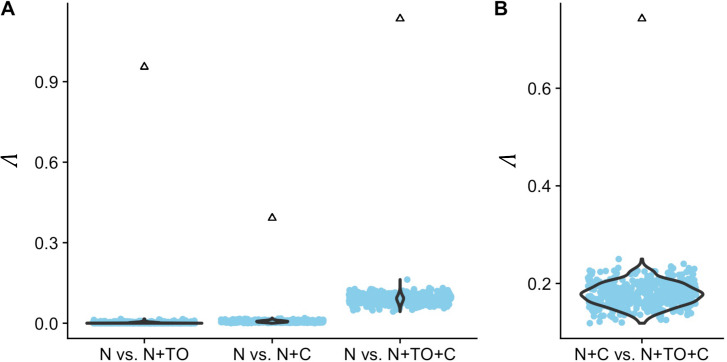
Comparative genomic approaches have been used to identify sites where mutations are under purifying selection and of functional consequence by searching for sequences that are conserved across distantly related species. However, the performance of these approaches has not been rigorously evaluated under population genetic models. Further, short-lived functional elements may not leave a footprint of sequence conservation across many species. We use simulations to study how one measure of conservation, the Genomic Evolutionary Rate Profiling (GERP) score, relates to the strength of selection (Nes). We show that the GERP score is related to the strength of purifying selection. However, changes in selection coefficients or functional elements over time (i.e. functional turnover) can strongly affect the GERP distribution, leading to unexpected relationships between GERP and Nes. Further, we show that for functional elements that have a high turnover rate, adding more species to the analysis does not necessarily increase statistical power. Finally, we use the distribution of GERP scores across the human genome to compare models with and without turnover of sites where mutations are under purifying selection. We show that mutations in 4.51% of the noncoding human genome are under purifying selection and that most of this sequence has likely experienced changes in selection coefficients throughout mammalian evolution. Our work reveals limitations to using comparative genomic approaches to identify deleterious mutations. Commonly used GERP score thresholds miss over half of the noncoding sites in the human genome where mutations are under purifying selection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Distribution and intensity of constraint in mammalian genomic sequence.Genome Res. 2005 Jul;15(7):901-13. doi: 10.1101/gr.3577405. Epub 2005 Jun 17. Genome Res. 2005. PMID: 15965027 Free PMC article.
-
Detection of nonneutral substitution rates on mammalian phylogenies.Genome Res. 2010 Jan;20(1):110-21. doi: 10.1101/gr.097857.109. Epub 2009 Oct 26. Genome Res. 2010. PMID: 19858363 Free PMC article.
-
Purifying selection maintains highly conserved noncoding sequences in Drosophila.Mol Biol Evol. 2007 Oct;24(10):2222-34. doi: 10.1093/molbev/msm150. Epub 2007 Jul 23. Mol Biol Evol. 2007. PMID: 17646256
-
What fraction of the human genome is functional?Genome Res. 2011 Nov;21(11):1769-76. doi: 10.1101/gr.116814.110. Epub 2011 Aug 29. Genome Res. 2011. PMID: 21875934 Free PMC article. Review.
-
Alignment-free phylogenetics and population genetics.Brief Bioinform. 2014 May;15(3):407-18. doi: 10.1093/bib/bbt083. Epub 2013 Nov 29. Brief Bioinform. 2014. PMID: 24291823 Review.
Cited by
-
Lineage Differentiation and Genomic Vulnerability in a Relict Tree From Subtropical Forests.Evol Appl. 2024 Nov 1;17(11):e70033. doi: 10.1111/eva.70033. eCollection 2024 Nov. Evol Appl. 2024. PMID: 39494192 Free PMC article.
-
Genic constraint against nonsynonymous variation across the mouse genome.BMC Genomics. 2023 Sep 22;24(1):562. doi: 10.1186/s12864-023-09637-2. BMC Genomics. 2023. PMID: 37736706 Free PMC article.
-
Exploring TTN variants as genetic insights into cardiomyopathy pathogenesis and potential emerging clues to molecular mechanisms in cardiomyopathies.Sci Rep. 2024 Mar 4;14(1):5313. doi: 10.1038/s41598-024-56154-7. Sci Rep. 2024. PMID: 38438525 Free PMC article. Review.
-
Isolation and Characterization of the Adamantinomatous Craniopharyngioma Primary Cells with Cancer-Associated Fibroblast Features.Biomedicines. 2025 Apr 9;13(4):912. doi: 10.3390/biomedicines13040912. Biomedicines. 2025. PMID: 40299526 Free PMC article.
-
High frequency of an otherwise rare phenotype in a small and isolated tiger population.Proc Natl Acad Sci U S A. 2021 Sep 28;118(39):e2025273118. doi: 10.1073/pnas.2025273118. Proc Natl Acad Sci U S A. 2021. PMID: 34518374 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
