

Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia
- PMID: 32471306
- PMCID: PMC7312673
- DOI: 10.3390/ijms21113810
Association of A Novel Splice Site Mutation in P/Q-Type Calcium Channels with Childhood Epilepsy and Late-Onset Slowly Progressive Non-Episodic Cerebellar Ataxia
Abstract
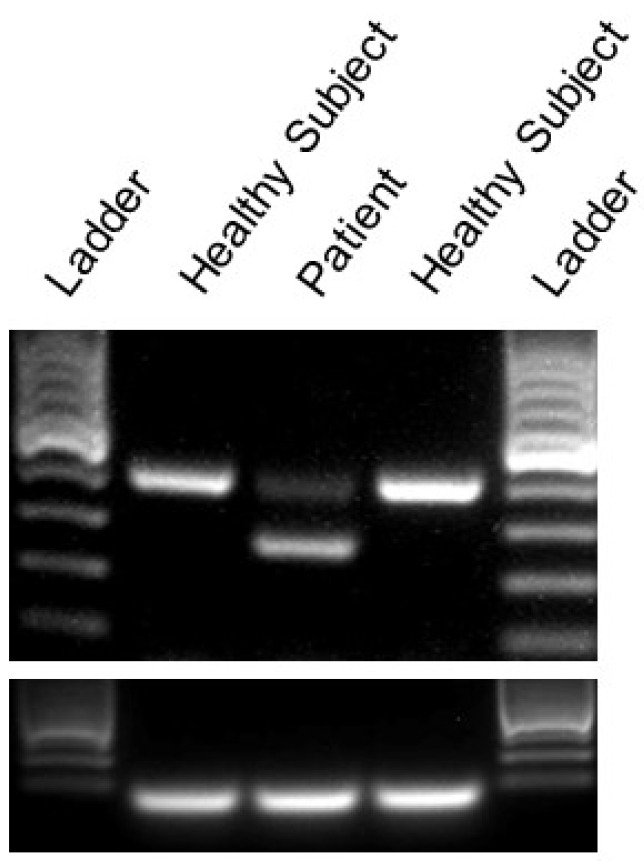
Episodic ataxia type 2 (EA2) is characterized by paroxysmal attacks of ataxia with typical onset in childhood or early adolescence. The disease is associated with mutations in the voltage-gated calcium channel alpha 1A subunit (Cav2.1) that is encoded by the CACNA1A gene. However, previously unrecognized atypical symptoms and the genetic overlap existing between EA2, spinocerebellar ataxia type 6, familial hemiplegic migraine type 1, and other neurological diseases blur the genotype/phenotype correlations, making a differential diagnosis difficult to formulate correctly and delaying early therapeutic intervention. Here we report a new clinical phenotype of a CACNA1A-associated disease characterized by absence epilepsy occurring during childhood. However, much later in life the patient displayed non-episodic, slowly progressive gait ataxia. Gene panel sequencing for hereditary ataxias led to the identification of a novel heterozygous CACNA1A mutation (c.1913 + 2T > G), altering the donor splice site of intron 14. This genetic defect was predicted to result in an in-frame deletion removing 44 amino acids from the voltage-gated calcium channel Cav2.1. An RT-PCR analysis of cDNA derived from patient skin fibroblasts confirmed the skipping of the entire exon 14. Furthermore, two-electrode voltage-clamp recordings performed from Xenopus laevis oocytes expressing a wild-type versus mutant channel showed that the genetic defect caused a complete loss of channel function. This represents the first description of distinct clinical manifestations that remarkably expand the genetic and phenotypic spectrum of CACNA1A-related diseases and should be considered for an early diagnosis and effective therapeutic intervention.
Keywords: CACNA1A mutation; P/Q-type calcium channel; absence epilepsy; cerebellar ataxia; next-generation sequencing.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures





References
-
- Haan J., Terwindt G.M., van den Maagdenberg A.M., Stam A.H., Ferrari M.D. A review of the genetic relation between migraine and epilepsy. Cephalalgia. 2008;28:105–113. - PubMed
-
- Damaj L., Lupien-Meilleur A., Lortie A., Riou E., Ospina L.H., Gagnon L., Vanasse C., Rossignol E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015;23:1505–1512. doi: 10.1038/ejhg.2015.21. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
