

A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity
- PMID: 32471981
- PMCID: PMC7260214
- DOI: 10.1038/s41467-020-16354-x
A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity
Abstract
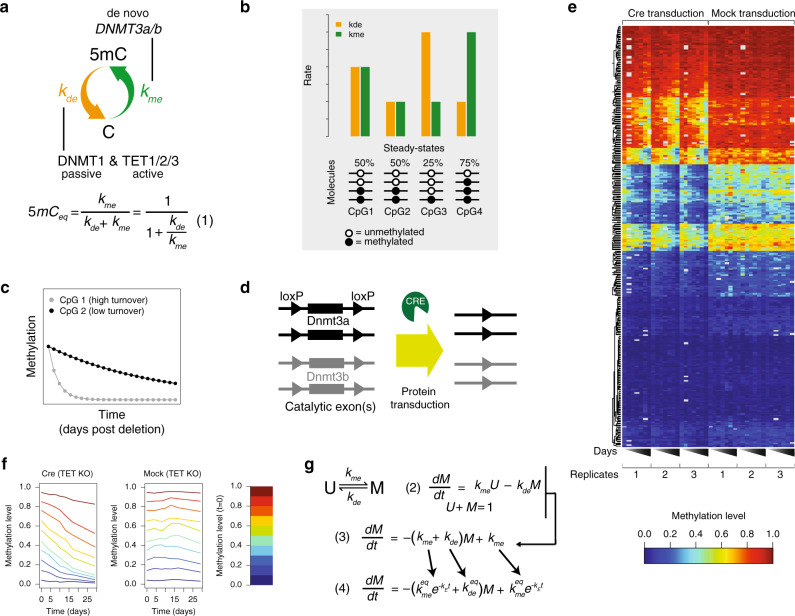
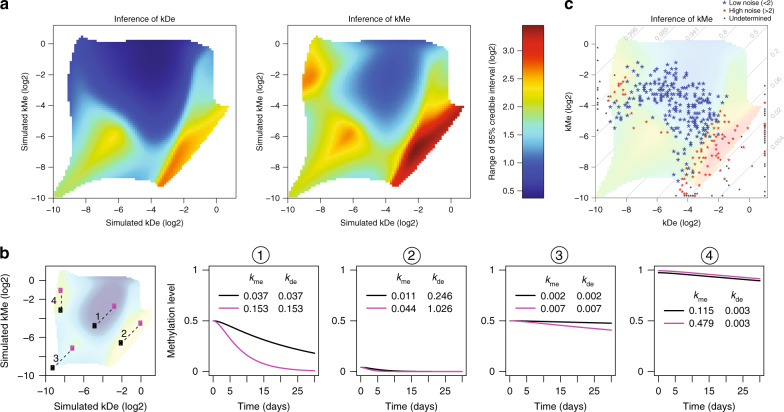
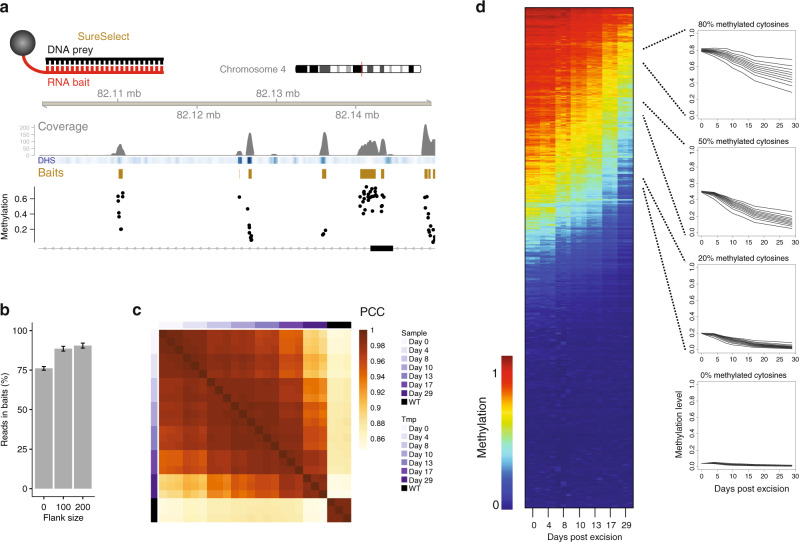
DNA methylation is considered a stable epigenetic mark, yet methylation patterns can vary during differentiation and in diseases such as cancer. Local levels of DNA methylation result from opposing enzymatic activities, the rates of which remain largely unknown. Here we developed a theoretical and experimental framework enabling us to infer methylation and demethylation rates at 860,404 CpGs in mouse embryonic stem cells. We find that enzymatic rates can vary as much as two orders of magnitude between CpGs with identical steady-state DNA methylation. Unexpectedly, de novo and maintenance methylation activity is reduced at transcription factor binding sites, while methylation turnover is elevated in transcribed gene bodies. Furthermore, we show that TET activity contributes substantially more than passive demethylation to establishing low methylation levels at distal enhancers. Taken together, our work unveils a genome-scale map of methylation kinetics, revealing highly variable and context-specific activity for the DNA methylation machinery.
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
The concurrence of DNA methylation and demethylation is associated with transcription regulation.Nat Commun. 2021 Sep 6;12(1):5285. doi: 10.1038/s41467-021-25521-7. Nat Commun. 2021. PMID: 34489442 Free PMC article.
-
DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells.Genome Biol. 2018 Jul 12;19(1):88. doi: 10.1186/s13059-018-1464-7. Genome Biol. 2018. PMID: 30001199 Free PMC article.
-
DNA methylation directs genomic localization of Mbd2 and Mbd3 in embryonic stem cells.Elife. 2016 Nov 16;5:e21964. doi: 10.7554/eLife.21964. Elife. 2016. PMID: 27849519 Free PMC article.
-
Active turnover of DNA methylation during cell fate decisions.Nat Rev Genet. 2021 Jan;22(1):59-66. doi: 10.1038/s41576-020-00287-8. Epub 2020 Oct 6. Nat Rev Genet. 2021. PMID: 33024290 Review.
-
Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase.Epigenetics. 2015;10(8):671-6. doi: 10.1080/15592294.2015.1062204. Epigenetics. 2015. PMID: 26098813 Free PMC article. Review.
Cited by
-
Temporally discordant chromatin accessibility and DNA demethylation define short and long-term enhancer regulation during cell fate specification.bioRxiv [Preprint]. 2024 Aug 27:2024.08.27.609789. doi: 10.1101/2024.08.27.609789. bioRxiv. 2024. Update in: Cell Rep. 2025 May 27;44(5):115680. doi: 10.1016/j.celrep.2025.115680. PMID: 39253426 Free PMC article. Updated. Preprint.
-
MeConcord: a new metric to quantitatively characterize DNA methylation heterogeneity across reads and CpG sites.Bioinformatics. 2022 Jun 24;38(Suppl 1):i307-i315. doi: 10.1093/bioinformatics/btac248. Bioinformatics. 2022. PMID: 35758820 Free PMC article.
-
The concurrence of DNA methylation and demethylation is associated with transcription regulation.Nat Commun. 2021 Sep 6;12(1):5285. doi: 10.1038/s41467-021-25521-7. Nat Commun. 2021. PMID: 34489442 Free PMC article.
-
TET3 dioxygenase modulates gene conversion at the avian immunoglobulin variable region via demethylation of non-CpG sites in pseudogene templates.Genes Cells. 2021 Mar;26(3):121-135. doi: 10.1111/gtc.12828. Epub 2021 Jan 31. Genes Cells. 2021. PMID: 33421268 Free PMC article.
-
DNA Methylation in the Adaptive Response to Exercise.Sports Med. 2024 Jun;54(6):1419-1458. doi: 10.1007/s40279-024-02011-6. Epub 2024 Apr 2. Sports Med. 2024. PMID: 38561436 Review.
References
-
- Baubec T, Schubeler D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet Dev. 2014;25:85–92. - PubMed
-
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33:245–254. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. - PubMed
-
- Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
