

Tumour predisposition and cancer syndromes as models to study gene-environment interactions
- PMID: 32472073
- PMCID: PMC8104546
- DOI: 10.1038/s41568-020-0265-y
Tumour predisposition and cancer syndromes as models to study gene-environment interactions
Abstract
Cell division and organismal development are exquisitely orchestrated and regulated processes. The dysregulation of the molecular mechanisms underlying these processes may cause cancer, a consequence of cell-intrinsic and/or cell-extrinsic events. Cellular DNA can be damaged by spontaneous hydrolysis, reactive oxygen species, aberrant cellular metabolism or other perturbations that cause DNA damage. Moreover, several environmental factors may damage the DNA, alter cellular metabolism or affect the ability of cells to interact with their microenvironment. While some environmental factors are well established as carcinogens, there remains a large knowledge gap of others owing to the difficulty in identifying them because of the typically long interval between carcinogen exposure and cancer diagnosis. DNA damage increases in cells harbouring mutations that impair their ability to correctly repair the DNA. Tumour predisposition syndromes in which cancers arise at an accelerated rate and in different organs - the equivalent of a sensitized background - provide a unique opportunity to examine how gene-environment interactions influence cancer risk when the initiating genetic defect responsible for malignancy is known. Understanding the molecular processes that are altered by specific germline mutations, environmental exposures and related mechanisms that promote cancer will allow the design of novel and effective preventive and therapeutic strategies.
Conflict of interest statement
Competing interests
S.T.A., B.B., A.B., W.C., J.E.C., C.M.C., W.D.F., G.G., J.L.G., E.P.H., P.M.H., T.W.M., D.M., F.N., declare no competing interests.
Figures
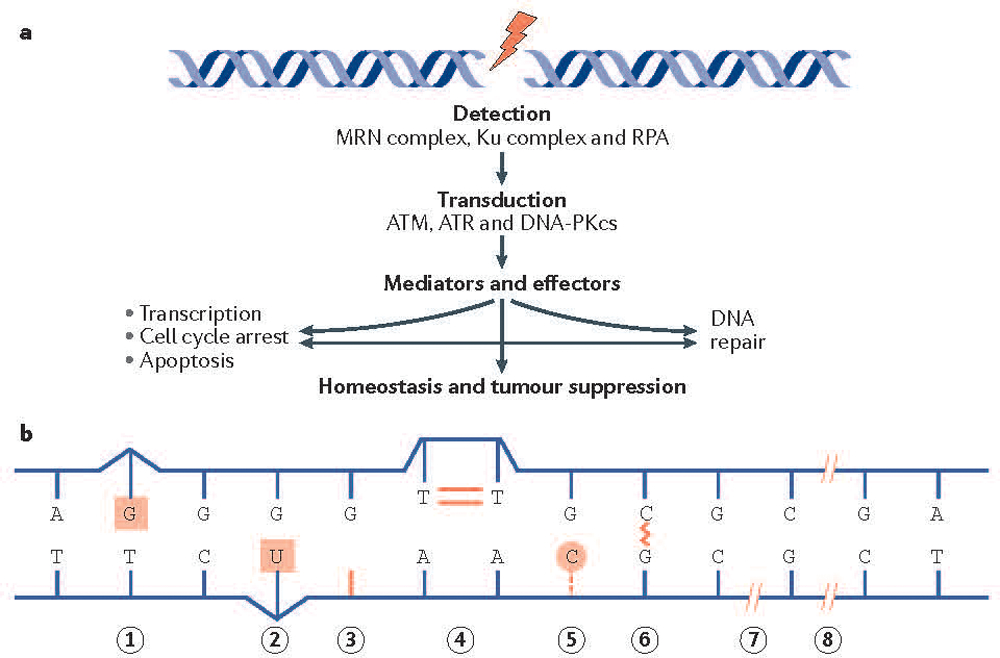
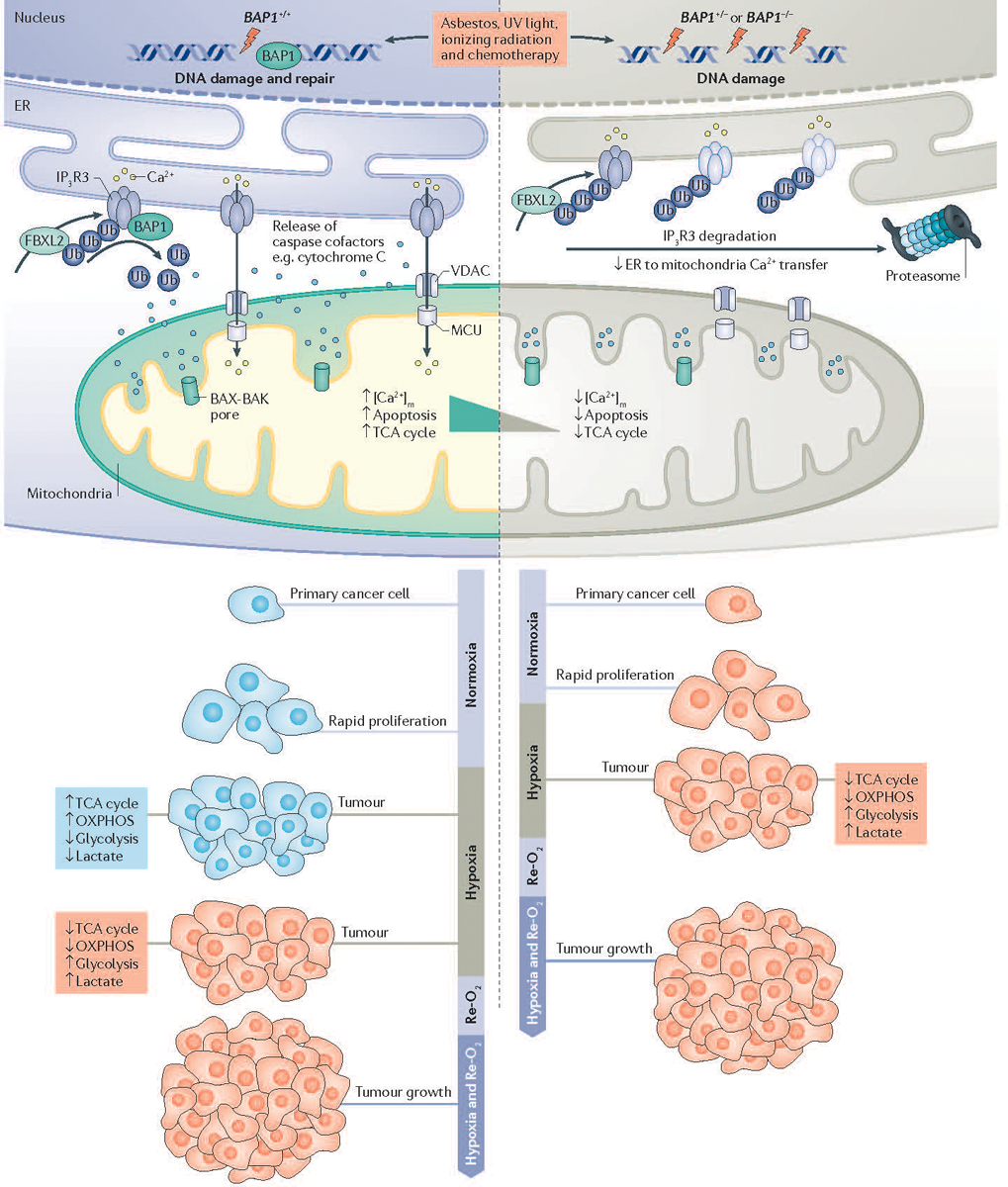
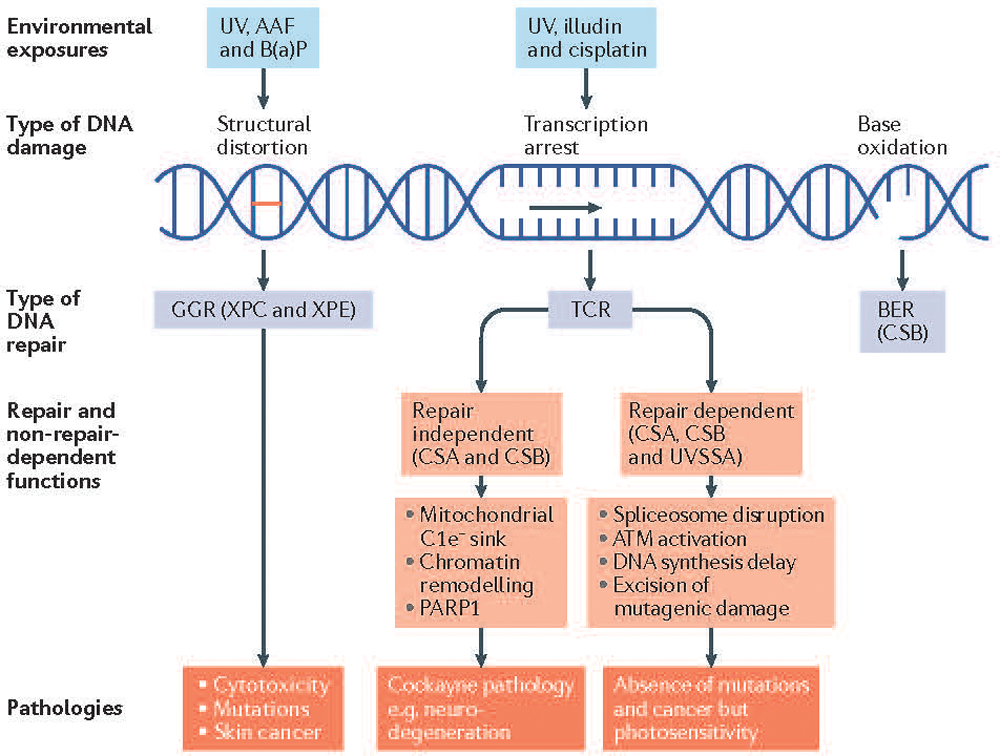
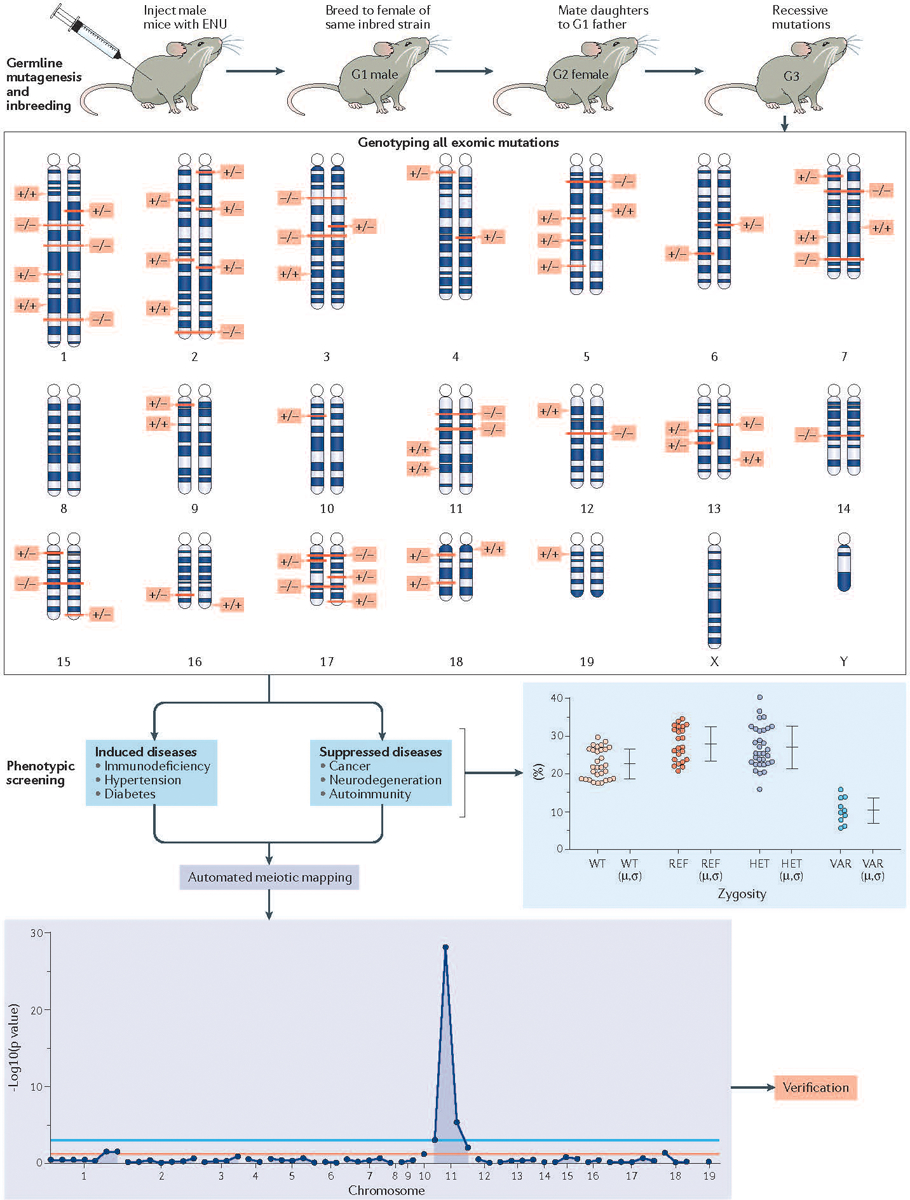
References
Publication types
MeSH terms
Grants and funding
- R01 CA198138/CA/NCI NIH HHS/United States
- R37 GM026017/GM/NIGMS NIH HHS/United States
- P01 CA077852/CA/NCI NIH HHS/United States
- R35 CA197706/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA237235/CA/NCI NIH HHS/United States
- R01 GM026017/GM/NIGMS NIH HHS/United States
- U01 CA111295/CA/NCI NIH HHS/United States
- U01 CA214195/CA/NCI NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 ES030948/ES/NIEHS NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 GM050006/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
