

Clinical and genetic characteristics of type I sialidosis patients in mainland China
- PMID: 32472645
- PMCID: PMC7318099
- DOI: 10.1002/acn3.51058
Clinical and genetic characteristics of type I sialidosis patients in mainland China
Abstract
Objective: Type I sialidosis (ST-1) is a rare autosomal recessive inherited disorder. To date, there has been no study on ST-1 patients in mainland China.
Methods: We reported in detail the cases of five Chinese ST-1 patients from two centers, and summarized all worldwide cases. Then, we compared the differences between Chinese and foreign patients.
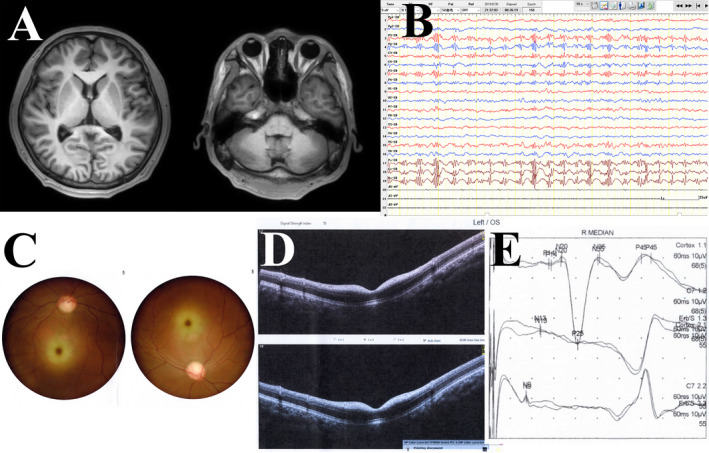
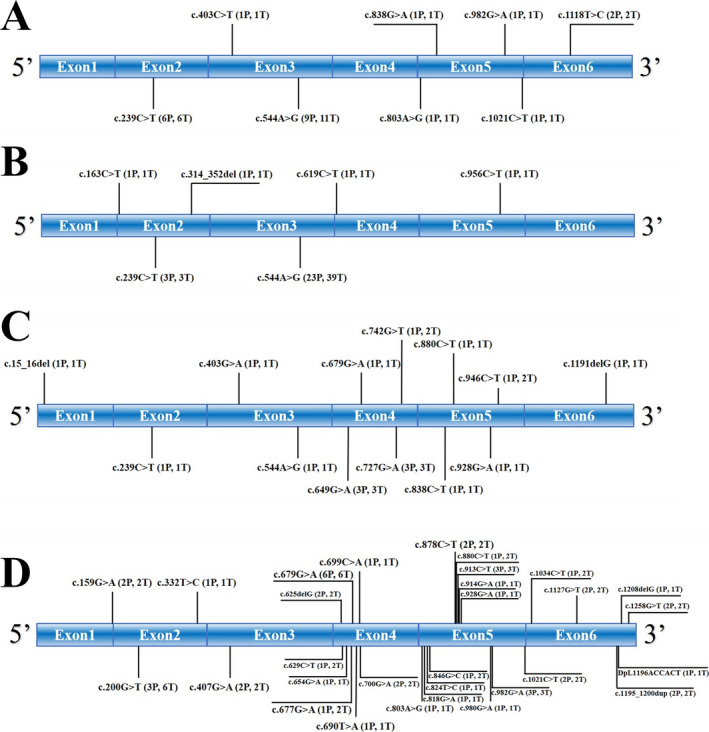
Results: A total of 77 genetically confirmed ST-1 patients were identified: 12 from mainland China, 23 from Taiwan, 10 from other Asian regions, and 32 from European and American regions. The mean age of onset was 16.0 ± 6.7 years; the most common symptoms were myoclonus seizures (96.0%), followed by ataxia (94.3%), and blurred vision (67.2%). Compared to other groups, the onset age of patients from mainland China was much younger (10.8 ± 2.7 years). The incidence of visual impairment was lower in patients from other Asian regions than in patients from mainland China and Taiwan (28.6% vs. 81.8%-100%). Cherry-red spots were less frequent in the Taiwanese patients than in patients from other regions (27.3% vs. 55.2%-90.0%). Furthermore, 48 different mutation types were identified. Chinese mainland and Taiwanese patients were more likely to carry the c.544A > G mutation (75% and 100%, respectively) than the patients from other regions (only 0%-10.0%). Approximately 50% of Chinese mainland patients carried the c.239C > T mutation, a much higher proportion than that found in the other populations. In addition, although the brain MRI of most patients was normal, 18 F-FDG-PET analysis could reveal cerebellar and occipital lobe hypometabolism.
Interpretation: ST-1 patients in different regions are likely to have different mutation types; environmental factors may influence clinical manifestations. Larger studies enrolling more patients are required.
© 2020 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
On behalf of all authors, the corresponding author confirms no conflict of interest.
Figures
References
-
- Cantz M, Gehler J, Spranger J. Mucolipidosis I: increased sialic acid content and deficiency of an alpha‐N‐acetylneuraminidase in cultured fibroblasts. Biochem Biophys Res Commun 1977;74:732–738. - PubMed
-
- Durand P, Gatti R, Cavalieri S, et al. Sialidosis (mucolipidosis I). Helv Paediatr Acta 1977;32:391–400. - PubMed
-
- Sphranger J, Gehler J, Cantz M. Mucolipidosis I–a sialidosis. Am J Med Genet 1977;1:21–29. - PubMed
-
- Franceschetti S, Canafoglia L. Sialidoses. Epileptic Disord 2016;18(S2):89–93. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
